Clear Sky Science · nl
Het gezamenlijke kennisbestand van ESM comprimeren in één eiwit-taalmodel
Waarom het samenbrengen van veel eiwitmodellen in één model ertoe doet
Iedere persoon draagt miljoenen kleine verschillen in zijn of haar DNA, waarvan veel single building blocks in eiwitten veranderen. De meeste van deze veranderingen zijn onschadelijk, maar sommige kunnen tot ziekte leiden. Artsen en onderzoekers willen graag snel en nauwkeurig kunnen bepalen welke veranderingen schadelijk zijn, alleen op basis van de eiwitsequentie. Dit artikel beschrijft een nieuwe strategie die de gezamenlijke kennis van vele bestaande eiwit-“taalmodellen” samenvat in één efficiënt systeem dat de huidige beste hulpmiddelen voor het beoordelen van de impact van genetische veranderingen evenaart of overtreft.
Van zinnen lezen naar eiwitten lezen
Eiwit-taalmodellen lenen ideeën van de technologie achter moderne vertaal- en chatsystemen. In plaats van woorden in een zin te leren, leren ze patronen in reeksen aminozuren, de letters van eiwitsequenties. Door te trainen op honderden miljoenen natuurlijke eiwitten pikken deze modellen signalen op over welke posities sterk geconserveerd zijn en welke verandering kunnen verdragen. Die patronen kunnen vervolgens worden gebruikt om genetische varianten te scoren: als een verandering een patroon doorbreekt dat de evolutie sterk beschermt, is de kans groter dat deze schadelijk is. Tot nu toe combineerden de krachtigste methoden deze modellen met extra informatie zoals 3D-structuur of evolutionaire stambomen, wat ze krachtig maar ook complex en soms moeilijk breed toepasbaar maakte.
Modellen elkaar laten leren
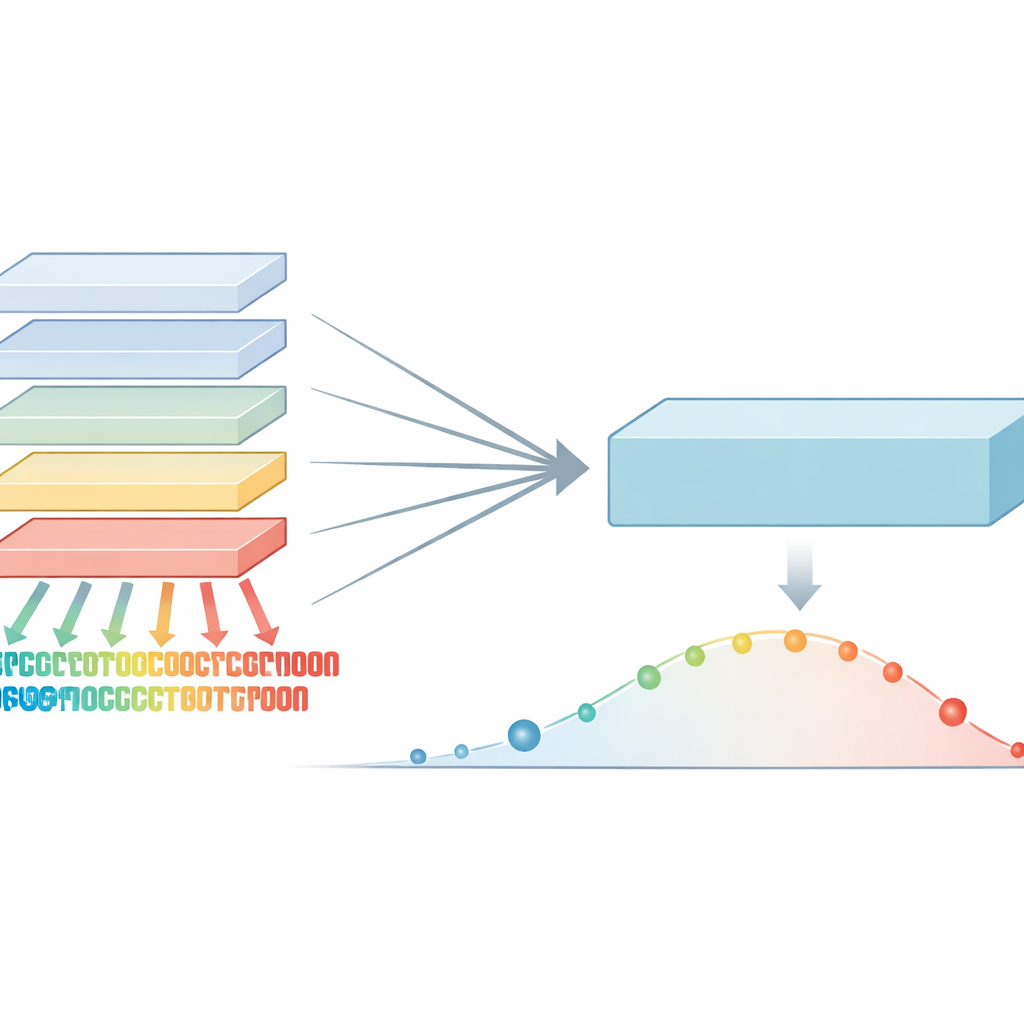
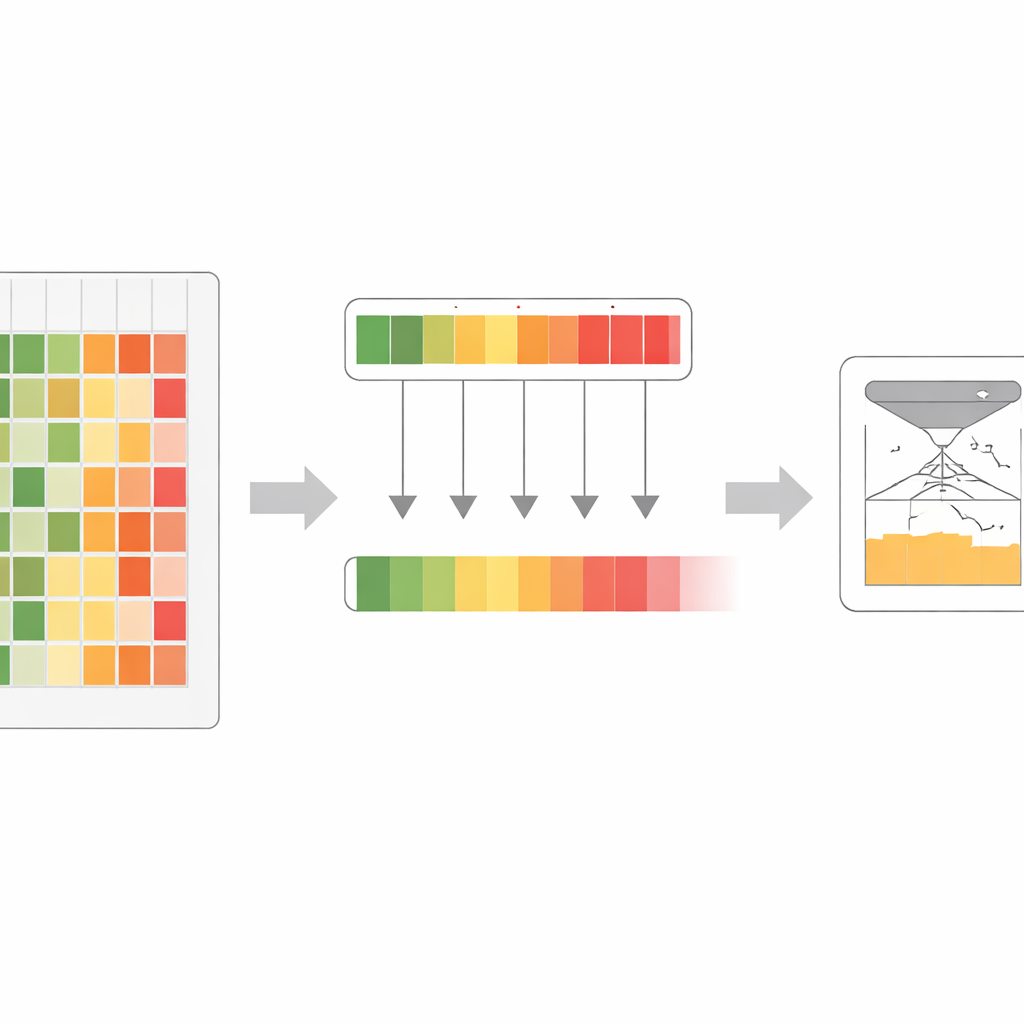
De auteurs merkten op dat verschillende eiwit-taalmodellen, zelfs wanneer ze op vergelijkbare gegevens zijn gebouwd, uiteenlopende sterke punten en blinde vlekken hebben. Het ene model kan bijvoorbeeld bijzonder scherp zijn in het signaleren van gevoelige regio’s in één eiwitfamilie, terwijl een ander elders uitblinkt. In plaats van hun meningen te middelen, introduceerde het team een “maximale-zekerheid”-regel: voor elke mogelijke eiwitverandering bekijken ze een hele familie modellen en behouden ze degene die het meest zeker is dat de verandering schadelijk is. Deze stap produceert een verrijkt set aan scores die de sterkste evolutionaire waarschuwingssignalen van elk model in de groep vastlegt, in plaats van ze te verdunnen.
Veel stemmen terugbrengen tot één
Met dit verrijkte signaal ontwierpen de onderzoekers een trainingsproces dat ze co-distillatie noemen. Hierbij oefenen alle oorspronkelijke modellen herhaaldelijk om het sterkste gecombineerde signaal te evenaren, waarbij ze beurtelings “student” en “leraar” zijn afhankelijk van waar elk model het meest zeker is. In vroege rondes helpt de maximale-zekerheid-regel om subtiele maar belangrijke patronen naar voren te halen die sommige modellen misten. In latere rondes zorgt een zachtere middelenstap ervoor dat de modellen het eens worden en ruis gladstrijken. Door meerdere cycli neemt één groot model geleidelijk bijna alle nuttige informatie over die eerder over het volledige ensemble verspreid was. Dit eindmodel, VESM-3B genaamd, wordt vervolgens gebruikt om kleinere versies te trainen die snel genoeg draaien voor grote genetische datasets terwijl ze het grootste deel van de nauwkeurigheid behouden.
Complexe systemen verslaan met alleen sequenties
Ondanks dat ze alleen ruwe eiwitsequenties als invoer gebruiken, evenaren of overtreffen de VESM-modellen verfijnde concurrenten die ook vertrouwen op 3D-structuren, handgebouwde evolutionaire geschiedenissen of populatiegegevens. Op klinische benchmarks afkomstig uit de ClinVar-database van ziektevarianten presteert het hoofdmodel van VESM beter dan veel veelgebruikte tools en doet het zelfs beter dan AlphaMissense, een recent hoogwaardig systeem dat structuur- en populatiegegevens combineert. Cruciaal is dat VESM’s prestaties standhouden voor zeer zeldzame varianten, de varianten die clinici het meest moeite kosten om te interpreteren. De modellen blinken ook uit in laboratoriummetingen die testen hoe mutaties eiwitfunctionaliteit, stabiliteit en binding beïnvloeden, en ze kunnen de grootte en richting van varianteffecten op echte klinische eigenschappen volgen in grote biobankdatasets.
Wat dit betekent voor genetica en geneeskunde
Door de sterke punten van vele sequentie-modellen zorgvuldig te combineren en te verfijnen, toont dit werk aan dat ruwe eiwitsequenties op zichzelf genoeg signaal kunnen bevatten om de impact van genetische veranderingen op state-of-the-art niveau te voorspellen. Het resultaat is een familie van hulpmiddelen die eenvoudiger te implementeren zijn dan systemen die zwaar op structuur leunen of veel data vereisen, en toch hoog nauwkeurig blijven voor eiwitten van mensen, microben en virussen. Voor clinici en onderzoekers betekent dit snellere, betrouwbaardere triage van DNA-varianten, een duidelijker beeld van hoe sterk een verandering een ziektegerelateerde eigenschap kan beïnvloeden, en een praktische manier om krachtige voorspellingsmodellen in dagelijkse genetica- en eiwitontwerpwerkstromen te brengen.
Bronvermelding: Dinh, T., Jang, SK., Zaitlen, N. et al. Compressing the collective knowledge of ESM into a single protein language model. Nat Methods 23, 772–784 (2026). https://doi.org/10.1038/s41592-026-03050-9
Trefwoorden: eiwit-taalmodellen, voorspelling van varianteffecten, genetische varianten, evolutionaire signalen, klinische genomica