Clear Sky Science · sv
XA-Novo: höggenomströmning masspektrometri‑baserad de novo‑sekvenseringsteknik för monoklonala antikroppar och antikroppsblandningar
Varför det är viktigt att avkoda antikroppar
Antikroppar är små Y‑formade proteiner som känner igen virus, bakterier och till och med cancerceller med anmärkningsvärd precision. För att omvandla dem till effektiva läkemedel eller diagnostiska verktyg behöver forskare deras exakta aminosyror—deras sekvens. Men att läsa av den sekvensen är ofta långsamt, dyrt och ibland omöjligt med nuvarande DNA‑baserade metoder. Denna studie presenterar XA‑Novo, en ny teknik som läser antikroppssekvenser direkt från proteinerna själva, med hjälp av masspektrometri och smarta algoritmer för att göra jobbet snabbare, mer exakt och även för komplexa blandningar av antikroppar.
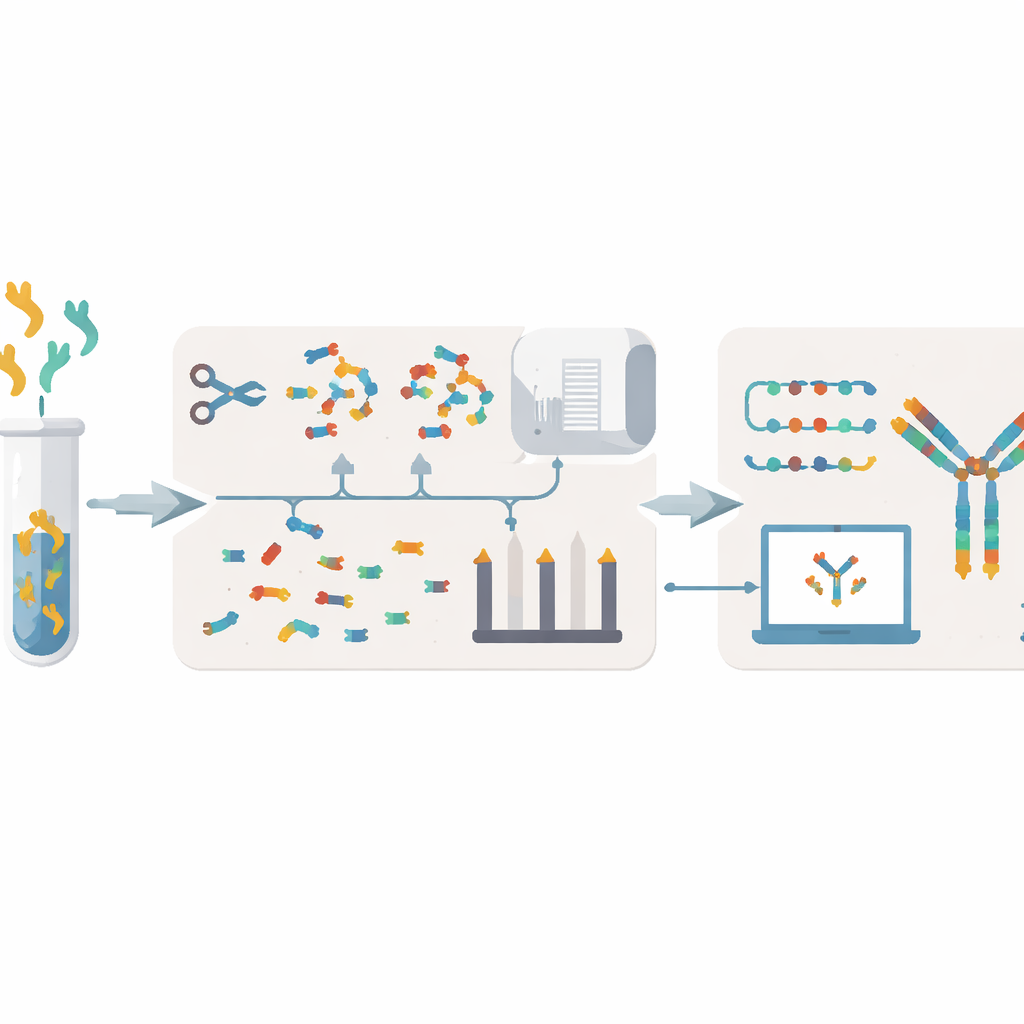
Nuvarande hinder för att läsa antikroppsrecept
Traditionella sätt att avkoda antikroppar utgår ofta från de celler som producerar dem. Forskare odlar hybridomceller eller isolerar B‑celler, extraherar deras genetiska material och sekvenserar därefter DNA eller RNA. Dessa tillvägagångssätt kan ta veckor till månader, kräver levande celler som kan vara känsliga eller förloras, och lämnar ibland luckor eller fel. De har också svårt att avgöra hur antikropparna som flyter i blod eller slem verkligen relaterar till B‑cellspopulationen som producerade dem. Ett alternativ är att arbeta på proteinnivå, att bryta ned antikroppar i mindre bitar och analysera dem med masspektrometri. Ändå kräver befintliga masspektrometrimetoder ofta stora mängder prov, har låg genomströmning och kan felmontera sekvenser, särskilt när många liknande antikroppar är närvarande samtidigt.
En ny pipeline som utgår från proteiner
XA‑Novo tar itu med dessa problem genom att kombinera förbättrad kemi, avancerad masspektrometri och modern maskininlärning i ett strömlinjeformat arbetsflöde. Först hackas antikropparna varsamt men grundligt till överlappande peptidfragment med en strategi som kallas "single‑pot multi‑enzymatic gradient digestion", där fem olika enzymer verkar i steg. Detta ökar fragmentens mångfald och överlappning utan att slösa dyrbart material. Därefter analyseras dessa fragment med högupplöst masspektrometri under två kompletterande fragmenteringslägen, vilket genererar rik spektralinformation som fångar hur varje peptid bryts upp.
Djupinlärning och smart sammansättning
När spektra har samlats in använder XA‑Novo en djupinlärningsmodell kallad Casanovo för att översätta de komplexa mönstren av masspek till förutsagda peptidsekvenser, ungefär som en språkmodell som översätter mellan språk. Dessa många korta "reads" skickas sedan till en ny assembler som heter Fusion. Fusion använder en beam‑search‑strategi och information från kända antikroppsramar för att sy ihop överlappande peptider till kompletta tunga och lätta kedjor. Den är utformad för att hantera vanliga problemområden—såsom aminosyror med nästan identiska massor och regioner där antikroppar varierar mest för bindning, kallade komplementaritetsbestämmande regioner—samt undvika luckor, insättningar och felordnade sträckor som kan förstöra funktionen.
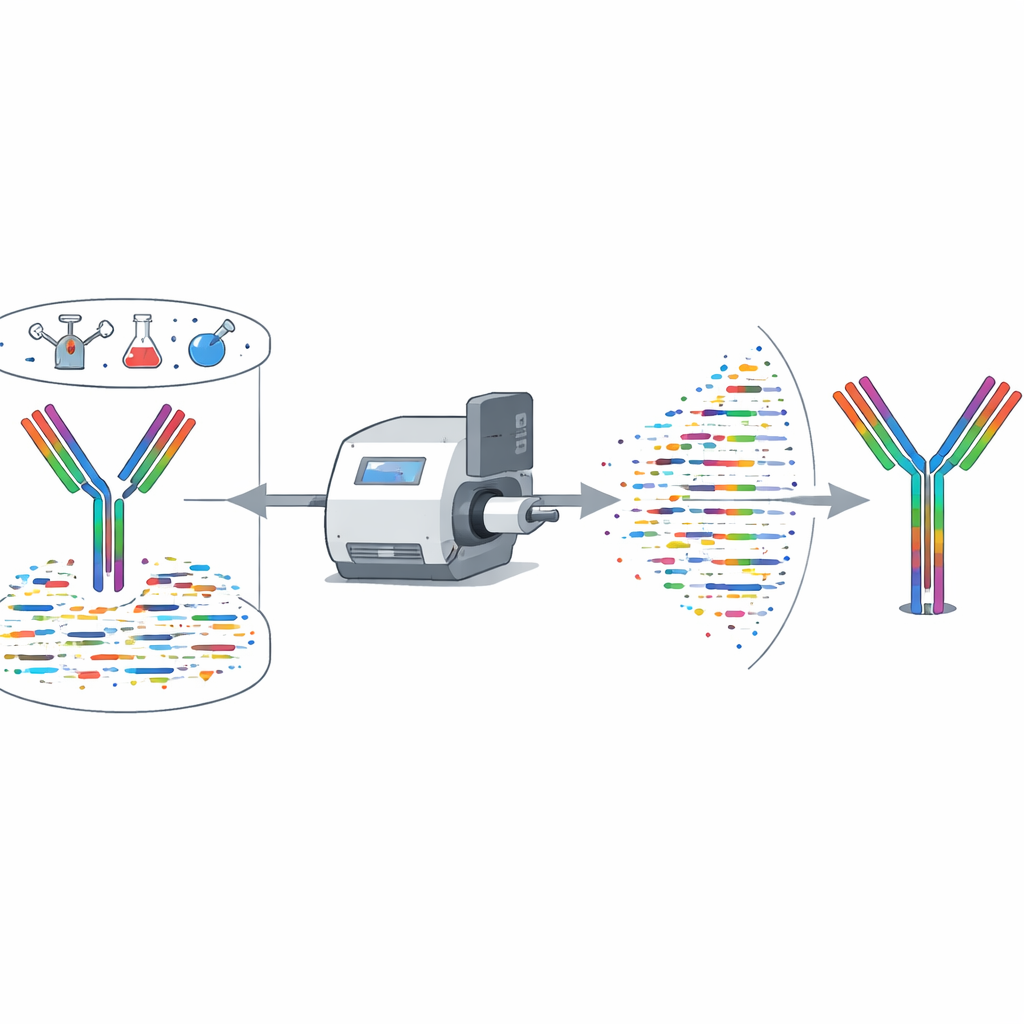
Att testa metoden
Författarna benchmarksatte noggrant XA‑Novo på antikroppar med kända sekvenser från människor och möss, inklusive flera som neutraliserar SARS‑CoV‑2. Jämfört med kommersiella verktyg och publika algoritmer uppnådde XA‑Novo konsekvent högre sekvenstäckning och noggrannhet, med kompletta och felfria rekonstruktioner över kritiska bindningsregioner. Den fungerade tillförlitligt även från så lite som 50 mikrogram antikropp. Teamet gav sig sedan på sex terapeutiska antikroppar vars sekvenser inte var offentligt tillgängliga. XA‑Novo avkodade deras tunga och lätta kedjor, sekvenserna klonades och uttrycktes, och de resulterande antikropparna testades i möss. In vivo‑experiment visade att dessa rekonstruerade antikroppar reducerade sina målceller eller makrofager lika effektivt som de ursprungliga kommersiella versionerna, vilket bekräftar att de avkodade sekvenserna var funktionellt korrekta.
Hantera antikroppsblandningar samtidigt
Många verkliga prover innehåller blandningar av antikroppar snarare än en enda ren komponent. XA‑Novo utmanades med blandningar av två eller tre COVID‑19‑neutraliserande antikroppar åt gången, för både mänskliga och musantikroppar. Systemet återvann varje komponents sekvens med minst 99,54 % korrekt täckning, ofta 100 %, inklusive de mest variabla bindningslooparna. Denna prestanda överträffar befintliga assemblers som vanligtvis är begränsade till enstaka antikroppar. Författarna byggde också ett webbaserat gränssnitt så att forskare kan ladda upp masspektrometriadatan och få rekonstruerade antikroppssekvenser och täckningskartor utan specialiserad hårdvara eller komplex installation.
Vad detta betyder för framtida antikroppsmediciner
XA‑Novo visar att det nu är möjligt att läsa ut fullständiga, mycket exakta antikroppssekvenser direkt från prov av proteiner, även i blandningar, med måttliga mängder material och ett till stor del automatiserat arbetsflöde. För icke‑specialister innebär detta att lovande antikroppar som upptäcks i labbet eller kliniken kan reverse‑engineeras snabbare, reproduceras pålitligt och vidareutvecklas till förbättrade varianter. Genom att göra antikroppssekvensering snabbare, mer skalbar och mindre beroende av känsliga cellinjer kan XA‑Novo påskynda grundläggande immunologistudier, hjälpa till att följa immunsvar vid infektioner som COVID‑19 och snabba på utvecklingen och optimeringen av antikroppsbaserade terapier.
Citering: Xiong, Y., Jiang, W., Xiao, J. et al. XA-Novo: high-throughput mass spectrometry-based de novo sequencing technology for monoclonal antibodies and antibody mixtures. Nat Commun 17, 3391 (2026). https://doi.org/10.1038/s41467-026-70496-y
Nyckelord: antikroppssekvensering, masspektrometri, monoklonala antikroppar, COVID-19 neutraliserande antikroppar, proteinengineering