Clear Sky Science · en
XA-Novo: high-throughput mass spectrometry-based de novo sequencing technology for monoclonal antibodies and antibody mixtures
Why decoding antibodies matters
Antibodies are tiny Y‑shaped proteins that recognize viruses, bacteria, and even cancer cells with remarkable precision. To turn them into powerful drugs or diagnostic tools, scientists need their exact amino‑acid "spelling"—their sequence. But reading that sequence is often slow, expensive, and sometimes impossible with current DNA‑based methods. This study introduces XA‑Novo, a new technology that reads antibody sequences directly from the proteins themselves, using mass spectrometry and smart algorithms to do the job faster, more accurately, and even for complex mixtures of antibodies.
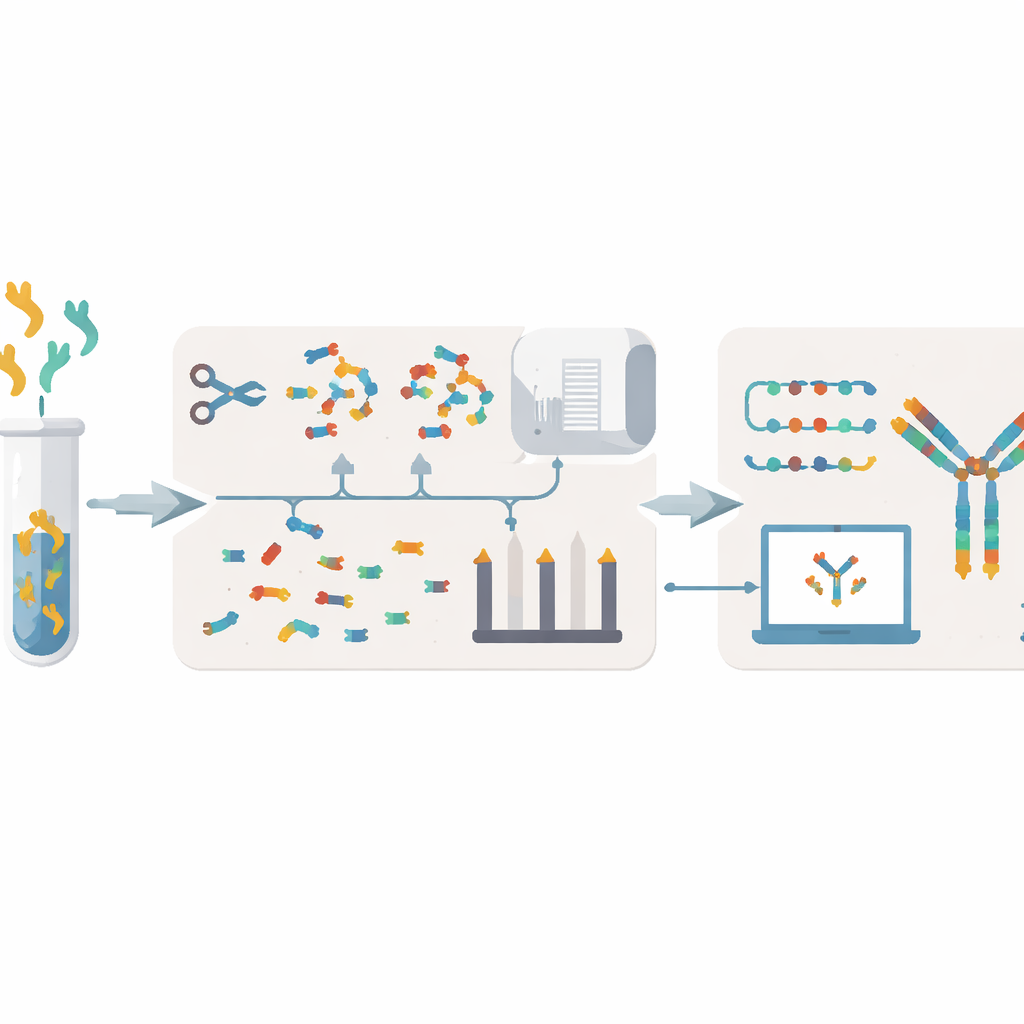
Current roadblocks to reading antibody recipes
Traditional ways of decoding antibodies usually start from the cells that produce them. Researchers grow hybridoma cells or isolate B cells, extract their genetic material, and then sequence the DNA or RNA. These approaches can take weeks to months, require living cells that may be fragile or lost, and sometimes still leave gaps or errors. They also struggle to tell how the antibodies floating in blood or mucus truly relate to the B‑cell population that produced them. An alternative is to work at the protein level, breaking antibodies into small pieces and analyzing them by mass spectrometry. Yet existing mass‑spectrometry methods often need large sample amounts, have low throughput, and can mis‑assemble sequences, especially when many similar antibodies are present together.
A new pipeline that starts from proteins
XA‑Novo tackles these issues by combining improved chemistry, advanced mass spectrometry, and modern machine learning into one streamlined workflow. First, antibodies are gently but thoroughly chopped into overlapping peptide fragments using a "single‑pot multi‑enzymatic gradient digestion" strategy, in which five different enzymes act in a time‑staggered way. This increases the diversity and overlap of fragments without wasting precious sample. Next, these fragments are analyzed by high‑resolution mass spectrometry under two complementary fragmentation modes, generating rich spectral information that captures how each peptide breaks apart.
Deep learning and smart assembly
Once spectra are collected, XA‑Novo uses a deep learning model called Casanovo to translate the complex patterns of mass peaks into predicted peptide sequences, much like a language model translating between languages. These many short "reads" are then passed to a new assembler named Fusion. Fusion uses a beam‑search strategy and information from known antibody templates to stitch overlapping peptides together into full heavy and light chains. It is designed to handle common problem spots—such as amino acids with nearly identical masses and regions where antibodies vary the most for binding, called complementarity‑determining regions—while avoiding gaps, insertions, and mis‑ordered stretches that can ruin function.
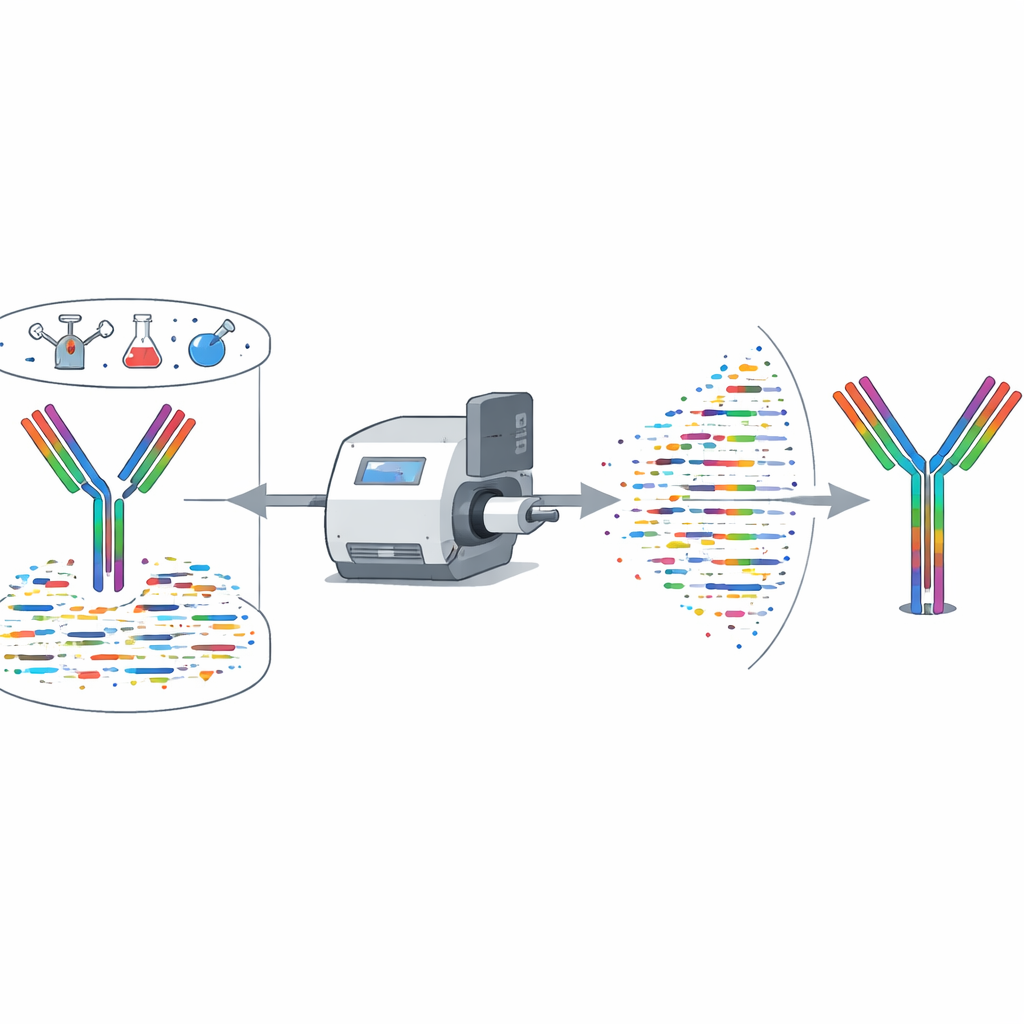
Putting the method to the test
The authors rigorously benchmarked XA‑Novo on antibodies with known sequences from humans and mice, including several that neutralize SARS‑CoV‑2. Compared to commercial tools and public algorithms, XA‑Novo consistently achieved higher sequence coverage and accuracy, with complete and error‑free reconstruction across critical binding regions. It worked reliably even when starting from as little as 50 micrograms of antibody. The team then tackled six therapeutic antibodies whose sequences were not publicly available. XA‑Novo decoded their heavy and light chains, the sequences were cloned and expressed, and the resulting antibodies were tested in mice. In vivo experiments showed that these reconstructed antibodies depleted their target immune cells or macrophages just as effectively as the original commercial versions, confirming that the decoded sequences were functionally correct.
Handling antibody mixtures at once
Many real‑world samples contain mixtures of antibodies rather than a single, pure one. XA‑Novo was challenged with blends of two or three COVID‑19 neutralizing antibodies at a time, for both human and mouse antibodies. The system recovered each component’s sequence with at least 99.54% accurate coverage, and often 100%, including the most variable binding loops. This performance surpasses existing assemblers that are typically limited to single antibodies. The authors also built a web‑based interface so researchers can upload mass‑spectrometry data and obtain reconstructed antibody sequences and coverage maps without specialized hardware or complex setup.
What this means for future antibody medicines
XA‑Novo shows that it is now possible to read out full, highly accurate antibody sequences directly from protein samples, even in mixtures, using modest amounts of material and a largely automated workflow. For non‑specialists, this means that promising antibodies discovered in the lab or clinic can be reverse‑engineered more quickly, reproduced reliably, and engineered into improved versions. By making antibody sequencing faster, more scalable, and less dependent on fragile cell lines, XA‑Novo could speed up basic immunology studies, help track immune responses to infections like COVID‑19, and accelerate the development and optimization of antibody‑based therapies.
Citation: Xiong, Y., Jiang, W., Xiao, J. et al. XA-Novo: high-throughput mass spectrometry-based de novo sequencing technology for monoclonal antibodies and antibody mixtures. Nat Commun 17, 3391 (2026). https://doi.org/10.1038/s41467-026-70496-y
Keywords: antibody sequencing, mass spectrometry, monoclonal antibodies, COVID-19 neutralizing antibodies, protein engineering