Clear Sky Science · pl
XA-Novo: technologia do de novo sekwencjonowania monoclonalnych przeciwciał i mieszanek przeciwciał oparta na wysokoprzepustowej spektrometrii mas
Dlaczego rozszyfrowanie przeciwciał ma znaczenie
Przeciwciała to niewielkie białka w kształcie litery Y, które rozpoznają wirusy, bakterie, a nawet komórki nowotworowe z imponującą precyzją. Aby przekształcić je w skuteczne leki lub narzędzia diagnostyczne, naukowcy potrzebują ich dokładnego „zapisu” aminokwasowego — sekwencji. Odczytanie tej sekwencji bywa jednak powolne, kosztowne i czasami niemożliwe przy użyciu obecnych metod opartych na DNA. W tym badaniu przedstawiono XA-Novo, nową technologię, która odczytuje sekwencje przeciwciał bezpośrednio z samych białek, wykorzystując spektrometrię mas i inteligentne algorytmy, dzięki czemu robi to szybciej, dokładniej i także dla złożonych mieszanek przeciwciał.
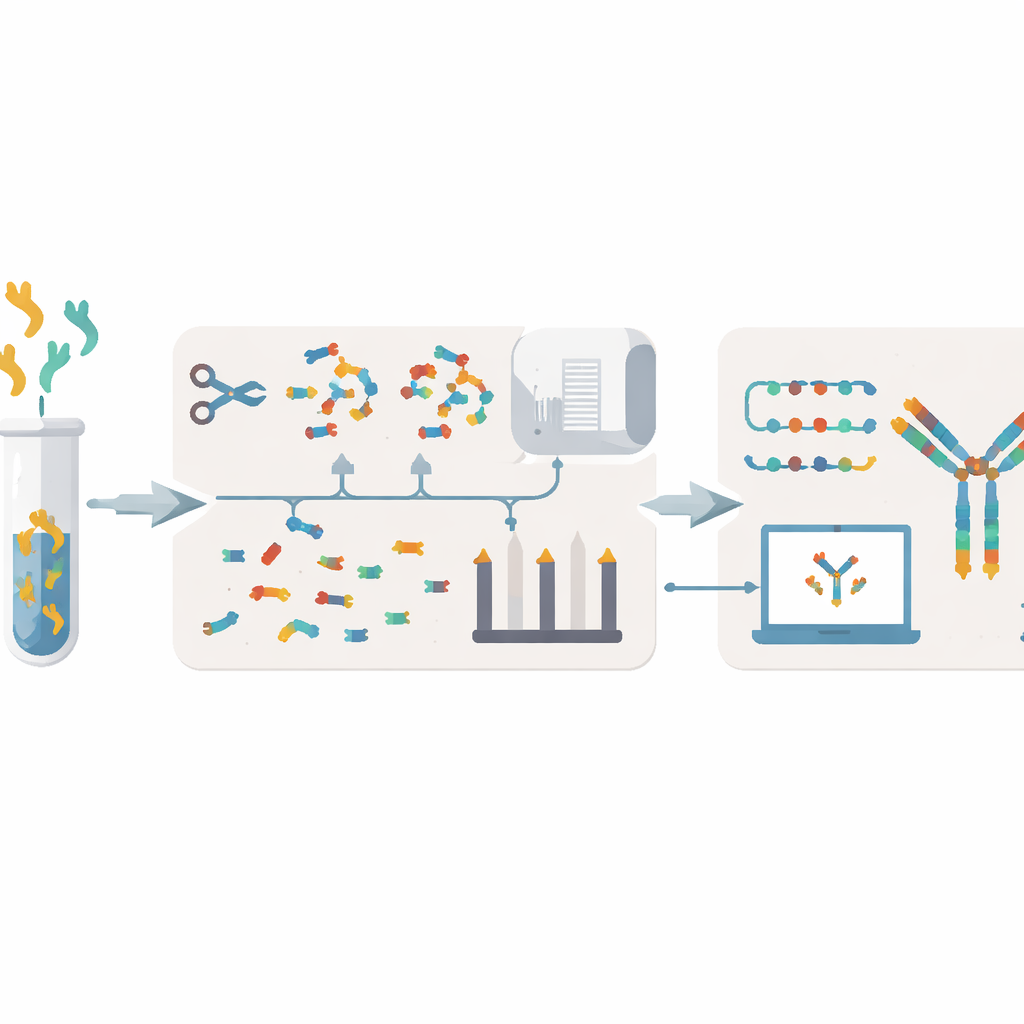
Obecne przeszkody w odczytywaniu „przepisów” na przeciwciała
Tradycyjne metody dekodowania przeciwciał zwykle zaczynają od komórek je produkujących. Badacze hodują hybrydomy lub izolują komórki B, ekstrahują materiał genetyczny, a następnie sekwencjonują DNA lub RNA. Podejścia te mogą trwać od tygodni do miesięcy, wymagają żywych komórek, które mogą być kruche lub utracone, i czasem pozostawiają luki lub błędy. Trudno im też określić, jak przeciwciała obecne we krwi czy śluzie faktycznie korelują z populacją komórek B, która je wyprodukowała. Alternatywą jest praca na poziomie białka, rozbijając przeciwciała na małe fragmenty i analizując je spektrometrią mas. Jednak istniejące metody spektrometrii często wymagają dużych ilości próbki, mają niską przepustowość i mogą błędnie składać sekwencje, zwłaszcza gdy obecnych jest wiele podobnych przeciwciał.
Nowy proces zaczynający się od białek
XA-Novo rozwiązuje te problemy, łącząc ulepszoną chemię, zaawansowaną spektrometrię mas i współczesne uczenie maszynowe w jeden usprawniony proces. Najpierw przeciwciała są delikatnie, a jednocześnie gruntownie rozcinane na nakładające się fragmenty peptydowe przy użyciu strategii „jednego naczynia, wieloenzymatycznego trawienia stopniowego”, w której pięć różnych enzymów działa w czasie przesuniętym względem siebie. Zwiększa to różnorodność i nakładanie się fragmentów bez marnowania cennej próbki. Następnie te fragmenty analizowane są przez spektrometrię mas wysokiej rozdzielczości w dwóch komplementarnych trybach fragmentacji, generując bogate widmowe dane opisujące, jak każdy peptyd rozpada się.
Uczenie głębokie i inteligentny montaż
Po zebraniu widm XA-Novo wykorzystuje model głębokiego uczenia o nazwie Casanovo do przetłumaczenia złożonych wzorców pików masowych na przewidywane sekwencje peptydowe, podobnie jak model językowy tłumaczy między językami. Te liczne krótkie „odczyty” trafiają następnie do nowego narzędzia montażowego o nazwie Fusion. Fusion używa strategii przeszukiwania wiązkowego (beam search) oraz informacji z znanych szablonów przeciwciał, aby zszyć nakładające się peptydy w pełne łańcuchy ciężkie i lekkie. Został zaprojektowany tak, by radzić sobie z powszechnymi problemami — takimi jak aminokwasy o niemal identycznej masie czy regiony o największej zmienności wiążącej (określane jako regiony determinujące komplementarność) — unikając jednocześnie luk, insercji i błędnych porządków, które mogą zniweczyć funkcję.
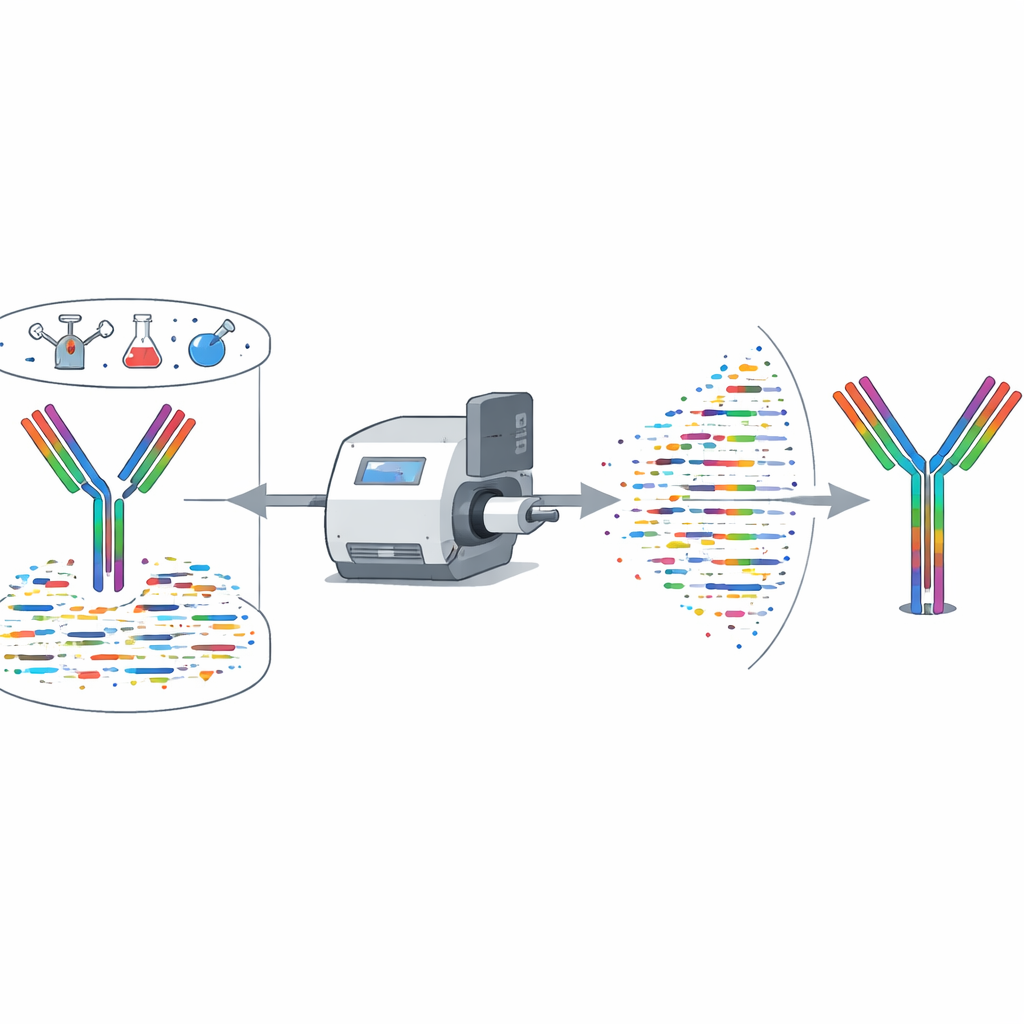
Weryfikacja metody
Autorzy rygorystycznie przetestowali XA-Novo na przeciwciałach o znanych sekwencjach pochodzących od ludzi i myszy, w tym kilku neutralizujących SARS-CoV-2. W porównaniu z narzędziami komercyjnymi i publicznymi algorytmami XA-Novo konsekwentnie osiągał wyższą pokrycie sekwencji i dokładność, z pełną i bezbłędną rekonstrukcją kluczowych regionów wiążących. Działał niezawodnie nawet przy rozpoczęciu z zaledwie 50 mikrogramami przeciwciała. Zespół następnie zmierzył się z sześcioma terapeutycznymi przeciwciałami, których sekwencje nie były publicznie dostępne. XA-Novo odczytał ich łańcuchy ciężkie i lekkie, sekwencje zostały sklonowane i wyrażone, a powstałe przeciwciała przetestowano na myszach. Eksperymenty in vivo wykazały, że zrekonstruowane przeciwciała zmniejszały liczebność docelowych komórek odpornościowych lub makrofagów tak samo skutecznie jak oryginalne komercyjne wersje, potwierdzając funkcjonalną poprawność odczytanych sekwencji.
Obsługa mieszanek przeciwciał jednocześnie
Wiele próbek z rzeczywistego świata zawiera mieszaniny przeciwciał, a nie pojedyncze czyste preparaty. XA-Novo zostało przetestowane na mieszankach dwóch lub trzech neutralizujących przeciwciał przeciw COVID-19 jednocześnie, dla przeciwciał ludzkich i mysich. System odtworzył sekwencje każdego składnika z co najmniej 99,54% dokładnym pokryciem, często osiągając 100%, włącznie z najbardziej zmiennymi pętlami wiążącymi. To osiągnięcie przewyższa istniejące narzędzia montażowe, które zwykle ograniczone są do pojedynczych przeciwciał. Autorzy uruchomili także interfejs webowy, dzięki któremu badacze mogą przesyłać dane ze spektrometrii mas i otrzymywać zrekonstruowane sekwencje przeciwciał oraz mapy pokrycia bez potrzeby posiadania wyspecjalizowanego sprzętu czy skomplikowanej konfiguracji.
Co to oznacza dla przyszłych leków przeciwciałowych
XA-Novo pokazuje, że możliwe jest teraz odczytanie pełnych, wysoce dokładnych sekwencji przeciwciał bezpośrednio z próbek białkowych, nawet w mieszaninach, używając umiarkowanych ilości materiału i w dużej mierze zautomatyzowanego procesu. Dla osób spoza specjalizacji oznacza to, że obiecujące przeciwciała odkryte w laboratorium lub klinice można szybciej odtworzyć, niezawodnie reprodukować i przekształcać w ulepszone wersje. Poprzez przyspieszenie, zwiększenie skali i zmniejszenie zależności od wrażliwych linii komórkowych, XA-Novo może przyspieszyć podstawowe badania immunologiczne, pomóc w śledzeniu odpowiedzi odpornościowej na infekcje takie jak COVID-19 oraz przyspieszyć rozwój i optymalizację terapii opartych na przeciwciałach.
Cytowanie: Xiong, Y., Jiang, W., Xiao, J. et al. XA-Novo: high-throughput mass spectrometry-based de novo sequencing technology for monoclonal antibodies and antibody mixtures. Nat Commun 17, 3391 (2026). https://doi.org/10.1038/s41467-026-70496-y
Słowa kluczowe: sekwencjonowanie przeciwciał, spektrometria mas, przeciwciała monoklonalne, neutralizujące przeciwciała przeciw COVID-19, inżynieria białek