Clear Sky Science · sv
En pardata språkmodell för modellering av protein–protein-interaktioner
Varför studera proteinpartnerskap är viktigt
Inne i varje cell arbetar proteiner sällan ensamma. De samarbetar i par eller större grupper för att föra vidare signaler, bygga strukturer och bekämpa infektioner. Att veta vilka proteiner som interagerar, hur hårt de binder och exakt var de vidrör varandra kan avslöja hur celler fungerar och hur sjukdomar uppstår. Men att experimentellt kartlägga alla dessa proteinpartnerskap i laboratoriet är både långsamt och kostsamt. Denna studie presenterar en ny artificiell intelligensmodell som läser par av proteinsekvenser tillsammans och lär sig att känna igen vem som binder vem, med vilken styrka och vid vilka kontaktpunkter — med enbart aminosyrasekvenser som indata.
Ett nytt sätt att läsa två proteiner samtidigt
De flesta nuvarande proteinspråkmodeller behandlar varje proteinkedja som om den vore ensam och bortser från hur den kan interagera med partner. Författarna byggde i stället en "Protein Pair Language Model" (PPLM) som alltid betraktar två sekvenser sida vid sida. Den använder en transformerarkitektur, en typ av djupinlärningsmodell som blivit vanlig inom språkteknologi, men anpassad för att separat följa mönster inom varje protein och mellan de två proteinerna. För att träna modellen sammanställde teamet över 3,3 miljoner proteinpar från strukturella databaser och interaktionsnätverk, vilket gav modellen en bred bild av hur naturliga proteiner tenderar att paras ihop i verklig biologi.
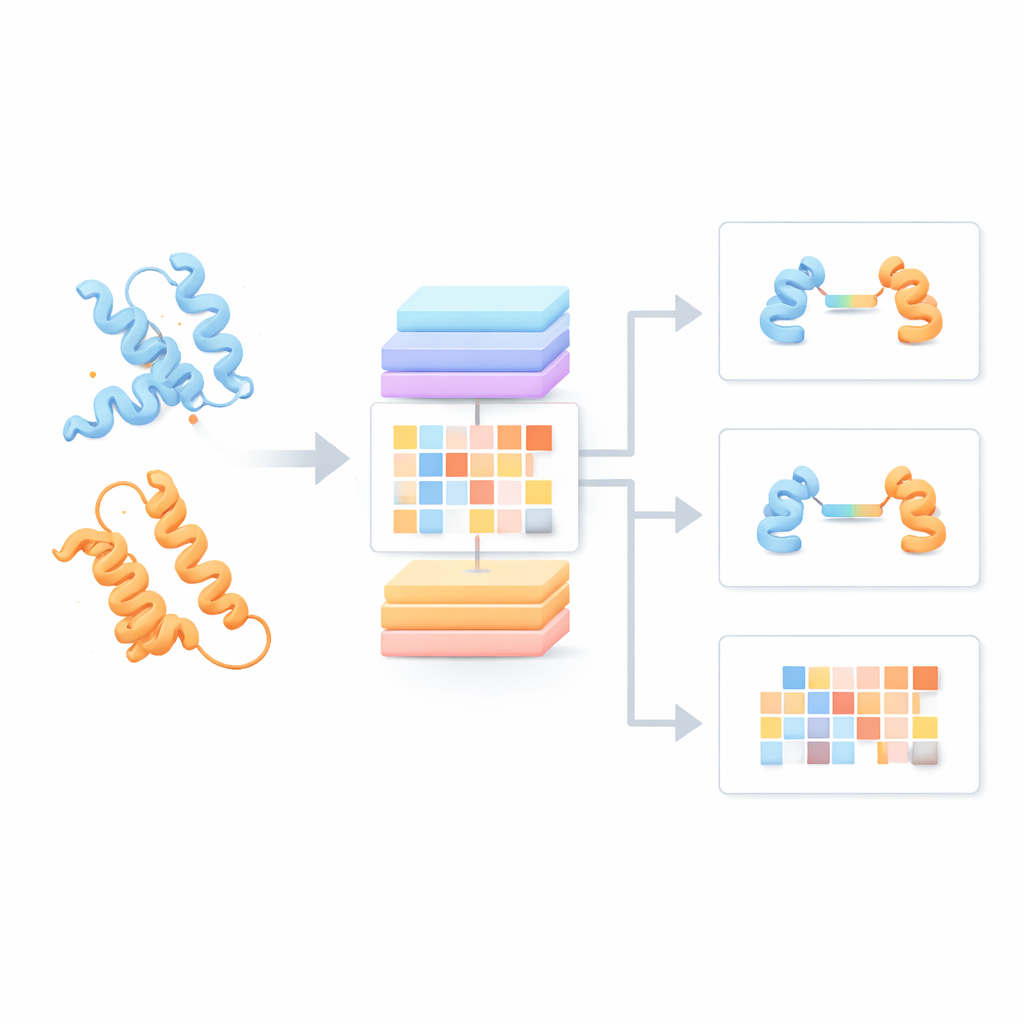
Att se interaktionssignaler dolda i sekvenser
För att testa om PPLM verkligen förstår proteinpartnerskap bad författarna modellen att förutsäga maskade aminosyror i sekvenspar och jämförde den med en ledande enkel-sekvensmodell kallad ESM2. Över tusentals proteinpar från olika källor var PPLM konsekvent mer säker och mer korrekt, särskilt vid residuer som ligger precis i gränssnittet där proteinerna berör varandra. Genom att visualisera modellens interna "attention"-mönster visade forskarna att PPLM naturligt fokuserar på dessa kontaktregioner, trots att den aldrig uttryckligen fått veta var gränssnittet ligger. I en detaljerad fallstudie av ett känt proteinkomplex överensstämde modellens starkast uppmärksammade residupar i stor utsträckning med de experimentellt observerade kontakterna i tredimensionellt utrymme.
Från grundläggande förståelse till praktiska prediktioner
Med denna grund skapade teamet tre tillämpade verktyg. PPLM-PPI förutspår om två proteiner sannolikt interagerar överhuvudtaget. Testad över fem olika arter överträffade den flera toppmoderna sekvensbaserade metoder och gav mer korrekta och stabila interaktionsbedömningar även när testproteiner skiljde sig mycket från dem som ingick i träningen. PPLM-Affinity uppskattar hur stark bindningen är mellan två proteiner. På en stor benchmark av komplex med uppmätta bindningsstyrkor slog den inte bara en version av ESM2 finjusterad för samma uppgift, utan överträffade även en specialiserad metod som använder detaljerade 3D-strukturer. Vinsterna var särskilt anmärkningsvärda för medicinskt viktiga system, såsom antikroppars bindning till antigen och T-cellreceptorer som känner igen immuntargets.
Att peka ut var proteinerna berör varandra
Det tredje verktyget, PPLM-Contact, zoomar in på vilka residupar över två proteiner som faktiskt kommer i kontakt. Det kombinerar PPLM:s cross-protein attention-mönster med evolutionär information från multipla sekvensaligneringar och distanskartor från individuella proteinstrukturer. Över flera utmanande testset återhämtade PPLM-Contact kontaktkartor och identifierade gränssnittsresiduier mer korrekt än befintliga metoder, inklusive sådana som i hög grad förlitar sig på strukturella indata. En förbättrad version, PPLM-Contact2, går ett steg längre genom att inkludera predikterade komplexstrukturer från moderna 3D-modelleringssystem. Detta hybridförfarande förbättrar kontaktprediktionen även utöver vad dessa strukturprediktorer själva kan åstadkomma, och erbjuder skarpare bilder av bindningsytor och mer tillförlitlig lokalisering av bindningsställen.
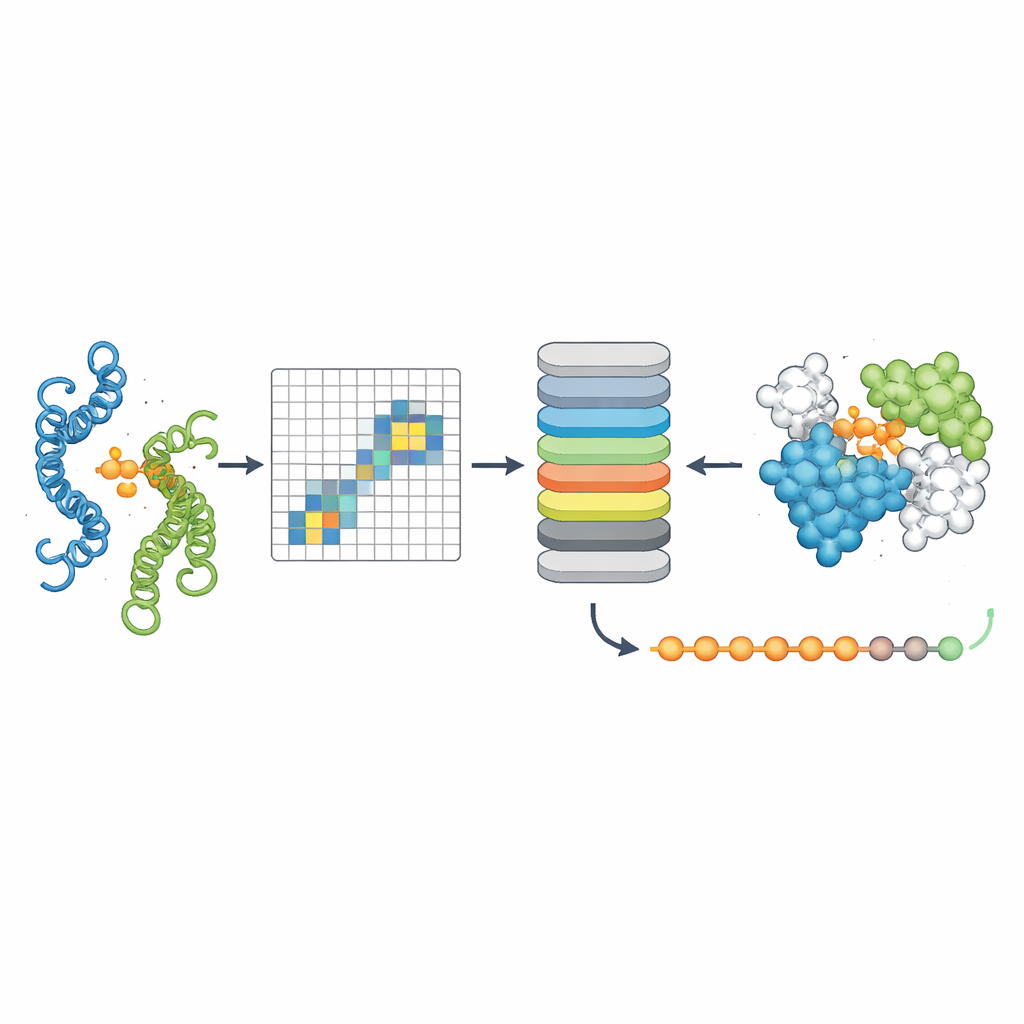
Vad detta betyder för biologi och medicin
Samlade visar resultaten att genom att läsa proteinsekvenser i par i stället för var för sig kan AI-modeller fånga subtila interaktionsmönster som ligger till grund för cellärt liv. PPLM och dess derivat kan säga om två proteiner sannolikt möts, hur hårt de greppar varandra och vilka aminosyror som utgör handslaget — allt från sekvensinformation som är billig och rikligt tillgänglig. Även om metoden fortfarande kämpar med mycket små eller svaga gränssnitt och är beroende av mångfalden i tillgängliga träningsdata, erbjuder den en skalbar väg mot att kartlägga interaktionsnätverk och vägleda designen av antikroppar, T-cellreceptorer och andra biologiska läkemedel. I huvudsak visar studien att samrepresenterade språkmodeller kan omvandla rå sekvensdata till rik, interaktionsmedveten insikt om hur proteiner samarbetar.
Citering: Liu, J., Chen, H. & Zhang, Y. A paired sequence language model for protein-protein interaction modeling. Nat Commun 17, 3733 (2026). https://doi.org/10.1038/s41467-026-70457-5
Nyckelord: protein–protein-interaktioner, proteinspråkmodeller, bindningsstyrka, prediktion av gränssnittskontakter, beräkningsbaserad strukturell biologi