Clear Sky Science · en
A paired sequence language model for protein-protein interaction modeling
Why studying protein partnerships matters
Inside every cell, proteins rarely work alone. They team up in pairs or larger groups to pass signals, build structures, and fight off infections. Knowing which proteins interact, how tightly they bind, and exactly where they touch can reveal how cells function and how diseases arise. But measuring all these protein partnerships in the lab is slow and expensive. This study introduces a new artificial intelligence model that reads pairs of protein sequences together, learning to recognize who binds whom, how strongly, and at what contact points—using only their amino-acid sequences as input.
A new way to read two proteins at once
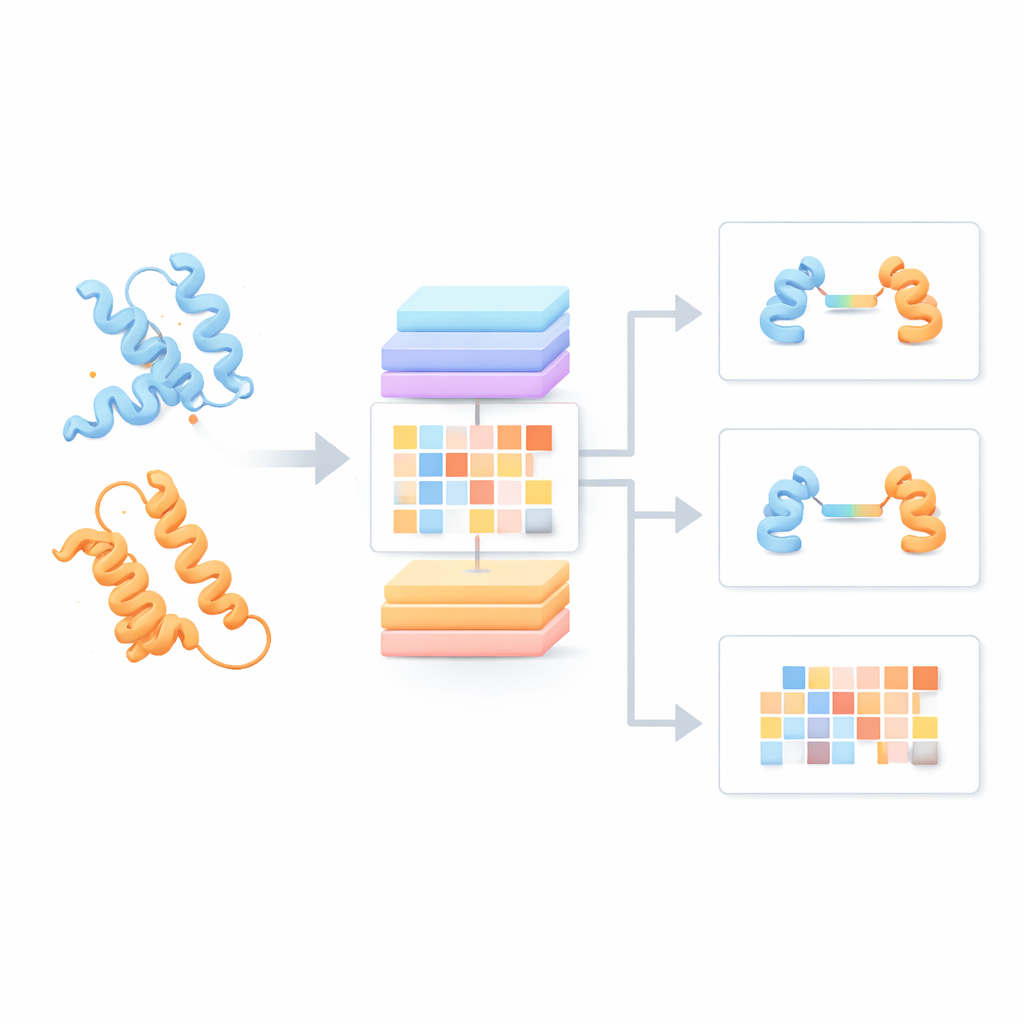
Most current protein language models treat each protein chain as if it were alone, ignoring how it might interact with partners. The authors instead built a "Protein Pair Language Model" (PPLM) that always looks at two sequences side by side. It uses a transformer architecture, a type of deep learning model popularized in language technologies, but customized so that it can separately track patterns within each protein and between the two proteins. To train it, the team assembled over 3.3 million protein pairs from structural databases and interaction networks, giving the model a broad view of how natural proteins tend to pair up in real biology.
Seeing interaction signals hidden in sequences
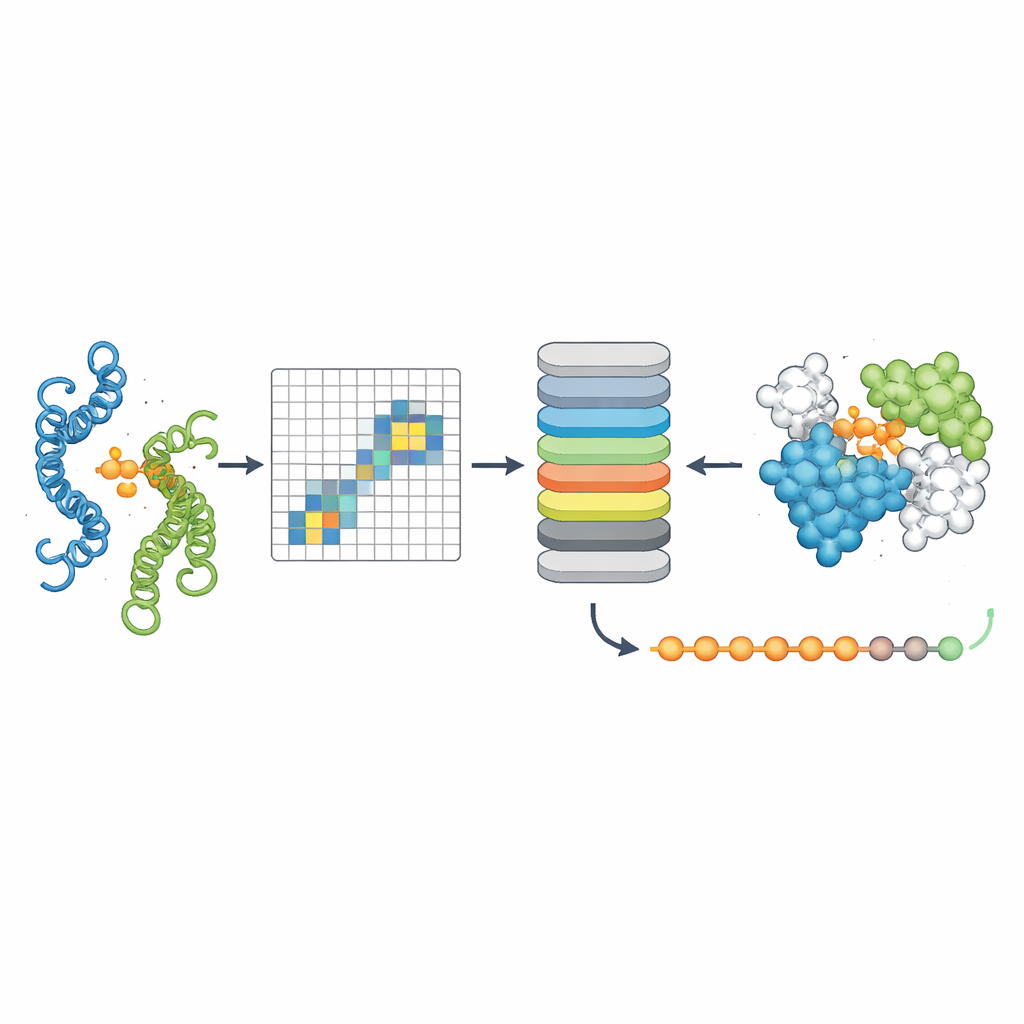
To test whether PPLM truly understands protein partnerships, the authors asked it to predict masked amino acids in sequence pairs and compared it to a leading single-sequence model called ESM2. Across thousands of protein pairs from different sources, PPLM was consistently more confident and accurate, especially at residues located right at the interface where the proteins touch. By visualizing the model’s internal "attention" patterns, the researchers showed that PPLM naturally focuses on those contact regions, even though it was never explicitly told where the interface lies. In a detailed case study of a known protein complex, the model’s most strongly attended residue pairs matched most of the experimentally observed contacts in three-dimensional space.
From basic understanding to practical predictions
Building on this foundation, the team created three applied tools. PPLM-PPI predicts whether two proteins are likely to interact at all. Tested across five different species, it outperformed several state-of-the-art sequence-based methods, delivering more accurate and more stable interaction calls even when test proteins were quite different from those seen in training. PPLM-Affinity estimates how strong the binding is between two proteins. On a large benchmark of complexes with measured binding strengths, it not only beat a version of ESM2 fine-tuned for the same task, but also surpassed a specialized method that uses detailed 3D structures. The gains were especially striking for medically important systems such as antibodies binding to antigens and T-cell receptors recognizing immune targets.
Pinpointing where proteins touch
The third tool, PPLM-Contact, zooms in on which residue pairs across two proteins actually make contact. It combines PPLM’s cross-protein attention patterns with evolutionary information from multiple sequence alignments and distance maps from individual protein structures. Across several challenging test sets, PPLM-Contact accurately recovered contact maps and identified interface residues better than existing methods, including some that rely heavily on structural inputs. An enhanced version, PPLM-Contact2, goes a step further by incorporating predicted complex structures from modern 3D modeling systems. This hybrid approach improves contact prediction even beyond those structure predictors themselves, offering sharper views of binding surfaces and more reliable localization of binding sites.
What this means for biology and medicine
Taken together, this work shows that reading protein sequences in pairs, rather than alone, allows AI models to capture subtle patterns of interaction that underlie cellular life. PPLM and its derivatives can say whether two proteins are likely to meet, how tightly they clasp, and which amino acids form the handshake—all from sequence information that is cheap and abundant. Although the approach still struggles with very small or weak interfaces and depends on the diversity of available training data, it offers a scalable path toward mapping interaction networks and guiding the design of antibodies, T-cell receptors, and other biologic drugs. In essence, the study demonstrates that co-represented language models can turn raw sequence data into rich, interaction-aware insight about how proteins work together.
Citation: Liu, J., Chen, H. & Zhang, Y. A paired sequence language model for protein-protein interaction modeling. Nat Commun 17, 3733 (2026). https://doi.org/10.1038/s41467-026-70457-5
Keywords: protein–protein interactions, protein language models, binding affinity, interface contact prediction, computational structural biology