Clear Sky Science · ru
Языковая модель парных последовательностей для моделирования белок–белковых взаимодействий
Почему важно изучать партнерства белков
В каждой клетке белки редко действуют в одиночку. Они объединяются в пары или более крупные комплексы, чтобы передавать сигналы, строить структуры и бороться с инфекциями. Знание того, какие белки взаимодействуют, насколько прочно они связываются и где именно соприкасаются, может раскрыть принципы работы клетки и механизмы развития заболеваний. Но экспериментальное измерение всех этих взаимодействий занимает много времени и дорого. В этом исследовании предлагается новая модель искусственного интеллекта, которая читает пары последовательностей белков вместе, обучаясь распознавать, кто с кем связывается, насколько сильно и в каких точках контакта — используя только аминокислотные последовательности как входные данные.
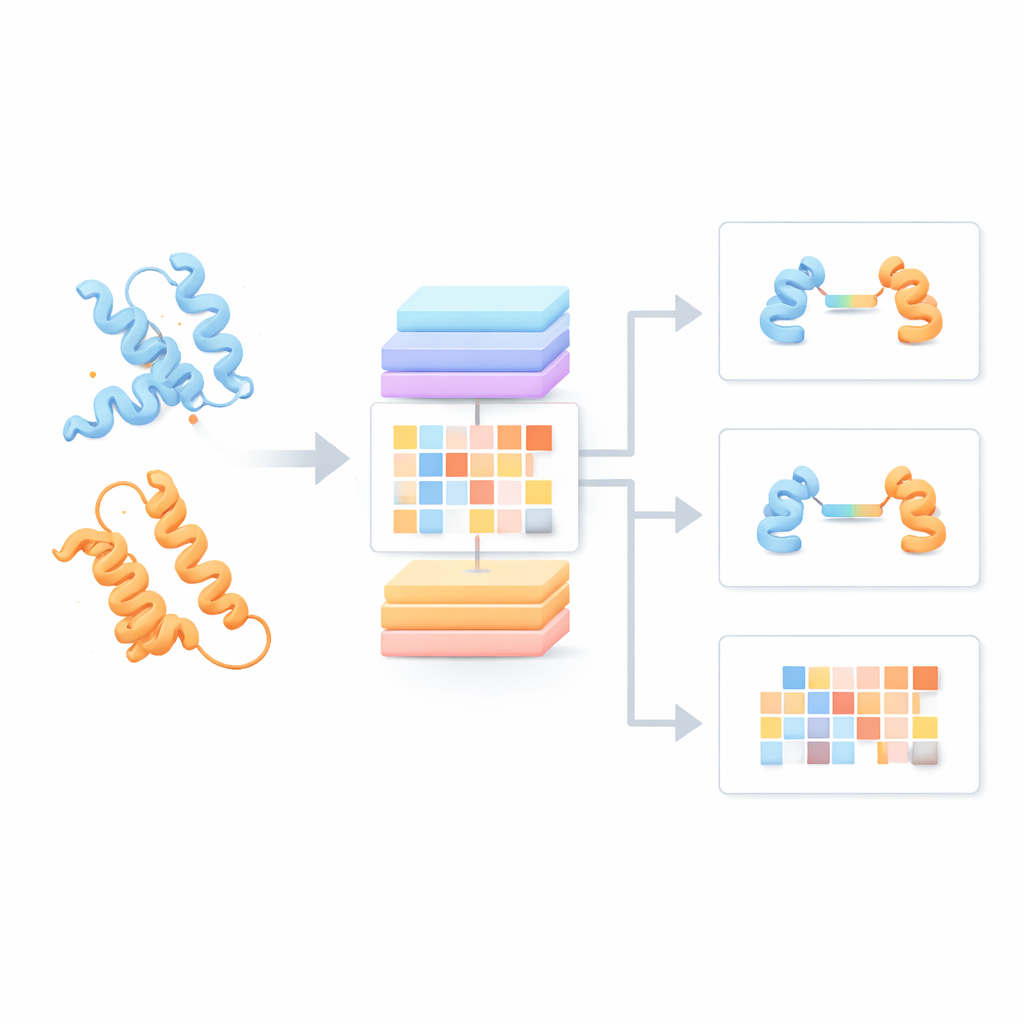
Новый способ одновременного чтения двух белков
Большинство современных языковых моделей для белков рассматривают каждую цепь как будто она одна, игнорируя возможное влияние партнеров. Авторы построили вместо этого «Языковую модель пар белков» (PPLM), которая всегда анализирует две последовательности рядом. Она использует архитектуру трансформера — тип глубокой нейросети, ставший популярным в языковых технологиях — но адаптированную так, чтобы отдельно отслеживать закономерности внутри каждой белковой цепи и между двумя цепями. Для обучения команда собрала более 3,3 миллиона пар белков из структурных баз данных и сетей взаимодействий, обеспечив модели широкий обзор природных закономерностей парообразования белков.
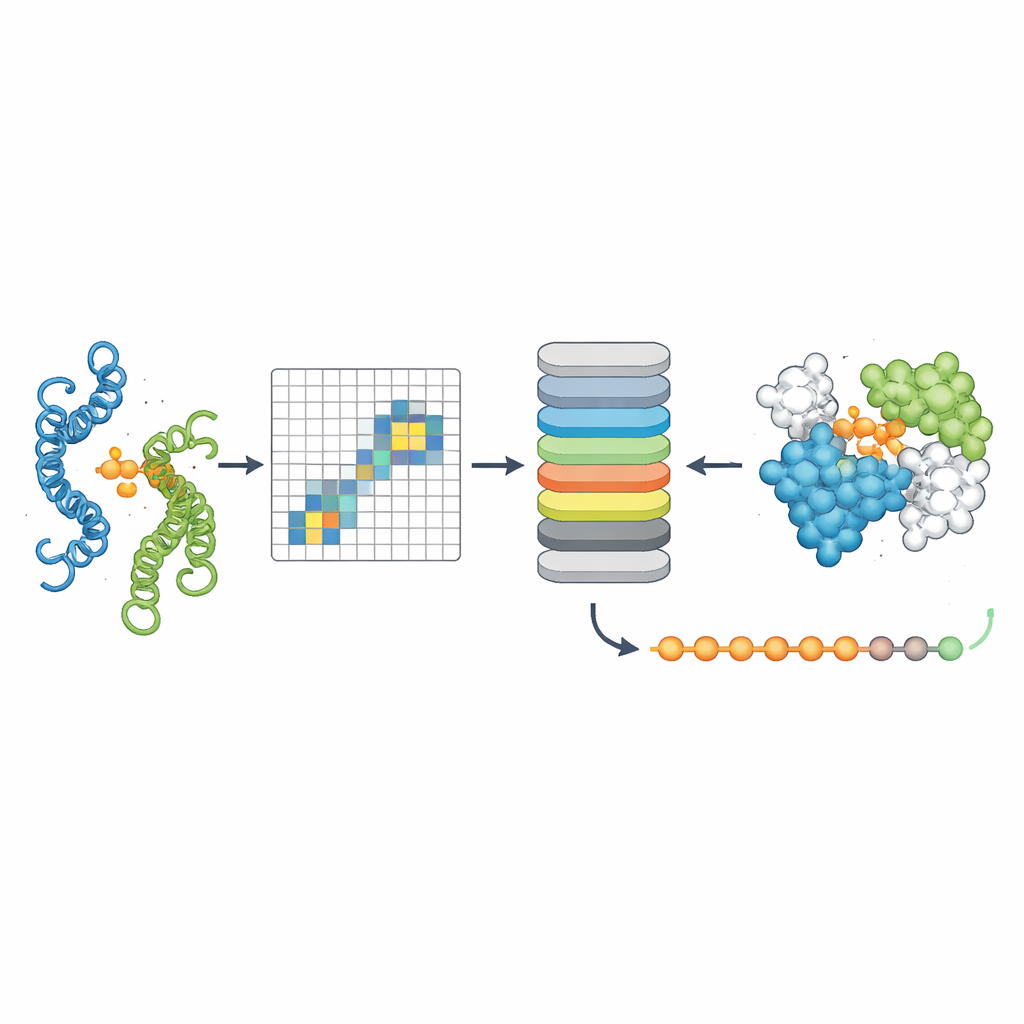
Видение сигналов взаимодействия, скрытых в последовательностях
Чтобы проверить, действительно ли PPLM понимает партнерства белков, авторы попросили модель предсказывать замаскированные аминокислоты в парных последовательностях и сравнили её с ведущей моделью для одиночных последовательностей ESM2. На тысячах пар белков из разных источников PPLM последовательно демонстрировала более высокую уверенность и точность, особенно в остатках, расположенных непосредственно в интерфейсе контакта. Визуализируя внутренние «внимательные» (attention) паттерны модели, исследователи показали, что PPLM естественно фокусируется на этих контактных областях, хотя ей никогда явно не указывали, где находится интерфейс. В детальном разборе известного белкового комплекса пары остатков с наибольшим вниманием модели совпадали с большинством экспериментально выявленных контактов в трехмерном пространстве.
От базового понимания к практическим предсказаниям
Опираясь на эту основу, команда создала три прикладных инструмента. PPLM-PPI предсказывает, вероятно ли вообще взаимодействие двух белков. Тестирование на пяти разных видах показало, что он превосходит несколько современных методов, основанных на последовательностях, обеспечивая более точные и более стабильные решения о взаимодействиях даже тогда, когда тестовые белки сильно отличаются от тех, что были в обучении. PPLM-Affinity оценивает силу связывания двух белков. На крупном бенчмарке комплексов с измеренной аффинностью она не только обошла версию ESM2, дообученную для той же задачи, но и превзошла специализированный метод, использующий детализированные 3D-структуры. Преимущества были особенно заметны для медицински важных систем, таких как антитела, связывающиеся с антигенами, и T-клеточные рецепторы, распознающие иммунные мишени.
Точное определение мест контакта белков
Третий инструмент, PPLM-Contact, фокусируется на том, какие пары остатков между двумя белками действительно вступают в контакт. Он комбинирует кросс-протеинные attention-паттерны PPLM с эволюционной информацией из множественных выравниваний последовательностей и картами расстояний из отдельных структур белков. На нескольких сложных тестовых наборах PPLM-Contact точно восстанавливал карты контактов и выявлял интерфейсные остатки лучше, чем существующие методы, включая некоторые, сильно зависящие от структурных входных данных. Улучшенная версия, PPLM-Contact2, идёт дальше, включая предсказанные структуры комплексов от современных 3D-моделирующих систем. Такой гибридный подход улучшает предсказание контактов даже по сравнению с самими предикторами структур, обеспечивая более чёткое представление поверхностей связывания и более надёжную локализацию сайтов связывания.
Что это значит для биологии и медицины
В совокупности работа показывает, что чтение последовательностей парно, а не по отдельности, позволяет ИИ-моделям захватывать тонкие закономерности взаимодействий, лежащие в основе клеточной жизни. PPLM и её производные могут сказать, вероятно ли встречаются два белка, насколько крепко они сцепляются и какие аминокислоты образуют «рукопожатие» — и всё это на основе дешёвой и обильной информации о последовательностях. Хотя подход по-прежнему испытывает трудности с очень малыми или слабыми интерфейсами и зависит от разнообразия доступных обучающих данных, он предлагает масштабируемый путь к картированию сетей взаимодействий и к направлению проектирования антител, T-клеточных рецепторов и других биологических препаратов. По сути, исследование демонстрирует, что совместно представленные языковые модели могут превратить сырые последовательностные данные в богатые, ориентированные на взаимодействия сведения о том, как белки работают вместе.
Цитирование: Liu, J., Chen, H. & Zhang, Y. A paired sequence language model for protein-protein interaction modeling. Nat Commun 17, 3733 (2026). https://doi.org/10.1038/s41467-026-70457-5
Ключевые слова: взаимодействия белок–белок, языковые модели белков, аффинность связывания, прогнозирование контактов интерфейса, вычислительная структурная биология