Clear Sky Science · pl
Model językowy dla sparowanych sekwencji w modelowaniu interakcji białko–białko
Dlaczego warto badać partnerskie relacje białek
W każdej komórce białka rzadko działają w pojedynkę. Tworzą pary lub większe zespoły, przekazując sygnały, budując struktury i zwalczając infekcje. Wiedza o tym, które białka wchodzą w interakcje, jak mocno się wiążą i w którym dokładnie miejscu stykają, może ujawnić mechanizmy funkcjonowania komórek i przyczyny chorób. Jednak eksperymentalne mierzenie wszystkich takich partnerskich relacji jest powolne i kosztowne. W tym badaniu wprowadzono nowy model sztucznej inteligencji, który analizuje pary sekwencji białkowych jednocześnie, ucząc się rozpoznawać, które białka się wiążą, jak silne jest to wiązanie i gdzie zachodzą punkty kontaktu — wykorzystując jedynie sekwencje aminokwasów jako dane wejściowe.
Nowy sposób czytania dwóch białek jednocześnie
Większość obecnych modeli językowych dla białek traktuje każdy łańcuch jako obiekt odizolowany, pomijając możliwe interakcje z partnerami. Autorzy zbudowali zamiast tego „Protein Pair Language Model” (PPLM), który zawsze analizuje dwie sekwencje obok siebie. Wykorzystuje on architekturę transformera, rodzaj głębokiego uczenia rozpowszechniony w technologiach językowych, ale dostosowaną tak, by odrębnie śledzić wzorce wewnątrz każdego białka oraz między dwoma białkami. Do treningu zespół zebrał ponad 3,3 miliona par białek z baz strukturalnych i sieci interakcji, dając modelowi szeroki przegląd tego, jak naturalnie łączą się białka w biologii.
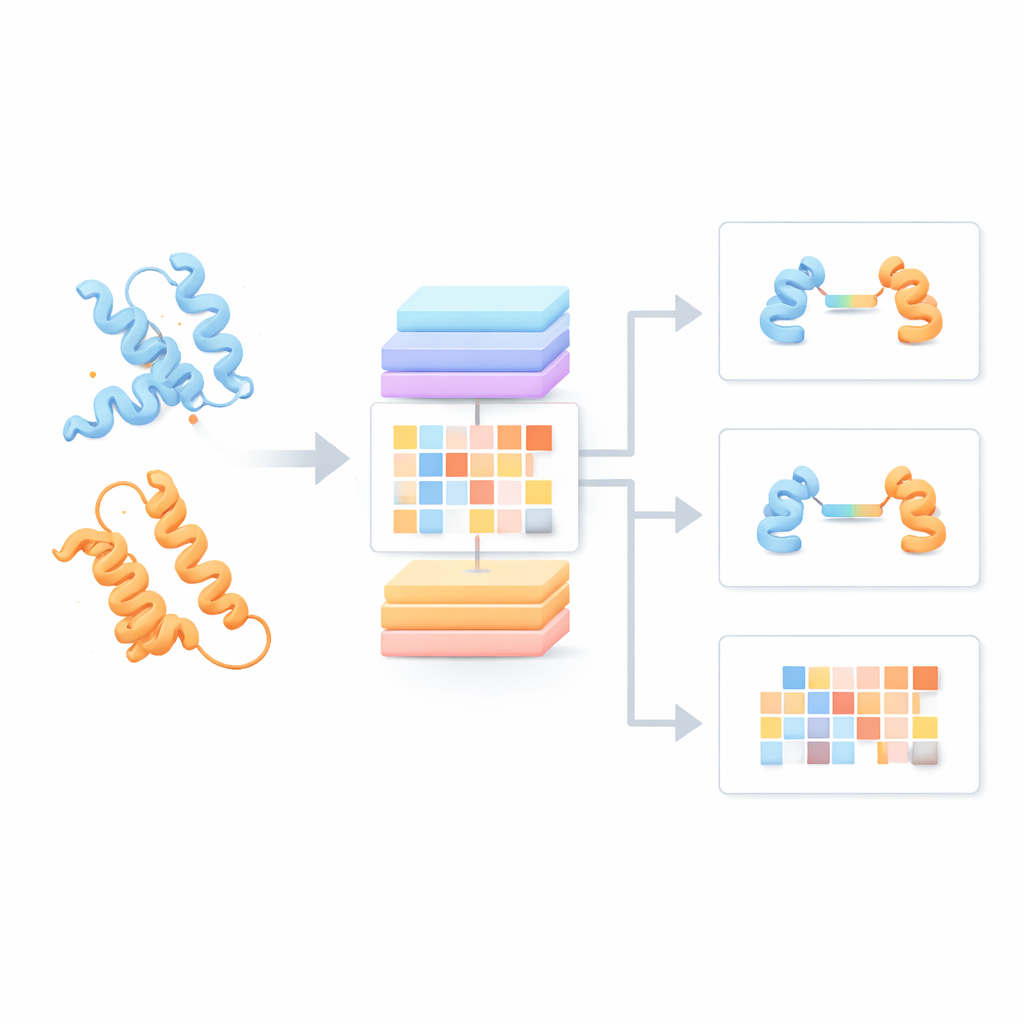
Odkrywanie sygnałów interakcji ukrytych w sekwencjach
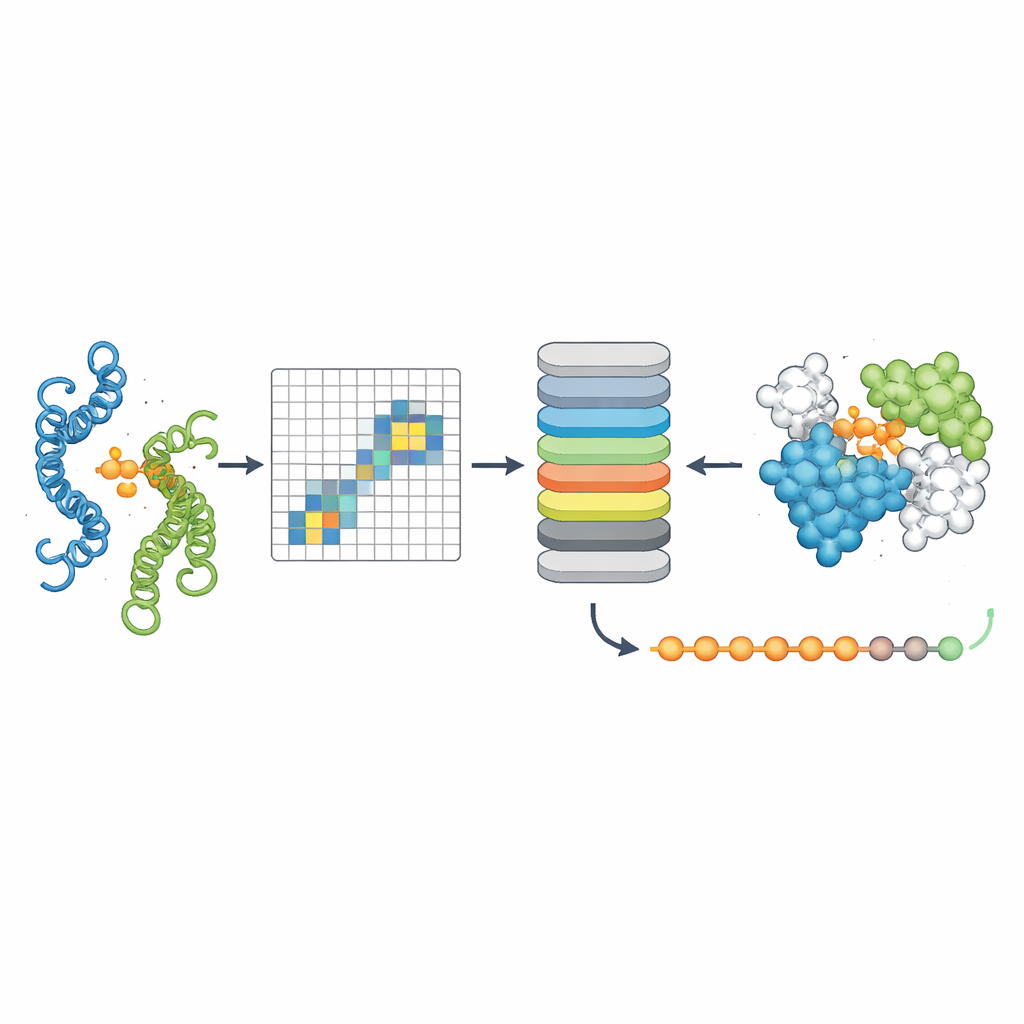
Aby sprawdzić, czy PPLM rzeczywiście rozumie partnerskie relacje białek, autorzy poprosili go o przewidywanie zamaskowanych aminokwasów w parach sekwencji i porównali go z wiodącym modelem pojedynczych sekwencji o nazwie ESM2. W testach obejmujących tysiące par białek z różnych źródeł PPLM był konsekwentnie bardziej pewny i dokładny, szczególnie w przypadku reszt położonych bezpośrednio w interfejsie, gdzie białka się stykają. Poprzez wizualizację wewnętrznych wzorców „uwagi” modelu badacze pokazali, że PPLM naturalnie skupia się na tych regionach kontaktu, mimo że nigdy nie poinformowano go explicite o lokalizacji interfejsu. W szczegółowym studium przypadku znanego kompleksu białkowego pary reszt z najsilniejszą uwagą modelu odpowiadały większości eksperymentalnie zaobserwowanych kontaktów w przestrzeni trójwymiarowej.
Od podstawowego rozumienia do praktycznych predykcji
W oparciu o te wyniki zespół stworzył trzy narzędzia stosowane w praktyce. PPLM-PPI przewiduje, czy dwie białka w ogóle mają prawdopodobieństwo interakcji. Testowany na pięciu różnych gatunkach przewyższał kilka nowoczesnych metod opartych na sekwencjach, dostarczając dokładniejszych i bardziej stabilnych decyzji o interakcji nawet gdy białka testowe znacznie różniły się od tych użytych w treningu. PPLM-Affinity szacuje, jak silne jest wiązanie między dwoma białkami. Na dużym zbiorze referencyjnym kompleksów z zmierzonymi siłami wiązania model nie tylko pokonał wersję ESM2 dostrojoną do tego zadania, lecz także przewyższył specjalistyczną metodę wykorzystującą szczegółowe struktury 3D. Zyski były szczególnie wyraźne dla systemów istotnych medycznie, takich jak przeciwciała wiążące antygeny czy receptory T-komórkowe rozpoznające cele immunologiczne.
Wskazywanie miejsca, gdzie białka się stykają
Trzecie narzędzie, PPLM-Contact, precyzuje, które pary reszt między dwoma białkami faktycznie nawiązują kontakt. Łączy ono wzorce uwagi PPLM między białkami z informacją ewolucyjną z wielokrotnych wyrównań sekwencji oraz mapami odległości pochodzącymi ze struktur poszczególnych białek. W kilku wymagających zestawach testowych PPLM-Contact dokładnie odtwarzał mapy kontaktów i lepiej identyfikował reszty interfejsu niż istniejące metody, w tym niektóre silnie polegające na danych strukturalnych. Udoskonalona wersja, PPLM-Contact2, idzie o krok dalej, integrując przewidywane struktury kompleksów z nowoczesnych systemów modelowania 3D. To podejście hybrydowe poprawia predykcję kontaktów nawet ponad samymi predyktorami struktur, oferując ostrzejszy obraz powierzchni wiązania i bardziej wiarygodną lokalizację miejsc wiążących.
Co to oznacza dla biologii i medycyny
Podsumowując, praca pokazuje, że czytanie sekwencji białkowych w parach, zamiast osobno, pozwala modelom AI wychwycić subtelne wzorce interakcji leżące u podstaw życia komórkowego. PPLM i jego pochodne potrafią określić, czy dwa białka prawdopodobnie się spotkają, jak mocno się ze sobą zwiążą i które aminokwasy tworzą „uścisk dłoni” — i to wszystko na podstawie sekwencji, która jest tania i powszechnie dostępna. Choć podejście wciąż ma trudności z bardzo małymi lub słabymi interfejsami i zależy od zróżnicowania dostępnych danych treningowych, oferuje skalowalną drogę do mapowania sieci interakcji oraz wspomagania projektowania przeciwciał, receptorów T-komórkowych i innych leków biologicznych. W istocie badanie pokazuje, że modele językowe współreprezentujące sekwencje mogą przekształcić surowe dane sekwencyjne w bogone, świadome interakcji wnioski o tym, jak białka współdziałają.
Cytowanie: Liu, J., Chen, H. & Zhang, Y. A paired sequence language model for protein-protein interaction modeling. Nat Commun 17, 3733 (2026). https://doi.org/10.1038/s41467-026-70457-5
Słowa kluczowe: interakcje białko–białko, modele językowe białek, siła wiązania, predykcja kontaktów w interfejsie, obliczeniowa biologia strukturalna