Clear Sky Science · es
Un modelo de lenguaje de secuencias pareadas para el modelado de interacciones proteína-proteína
Por qué importa estudiar las colaboraciones entre proteínas
Dentro de cada célula, las proteínas rara vez actúan solas. Se asocian en pares o en complejos mayores para transmitir señales, construir estructuras y combatir infecciones. Conocer qué proteínas interactúan, con qué fuerza se unen y exactamente dónde se tocan puede revelar cómo funcionan las células y cómo surgen las enfermedades. Pero medir todas estas asociaciones de proteínas en el laboratorio es lento y costoso. Este estudio presenta un nuevo modelo de inteligencia artificial que lee pares de secuencias proteicas juntos, aprendiendo a reconocer quién se une a quién, con qué intensidad y en qué puntos de contacto —usando solo sus secuencias de aminoácidos como entrada.
Una nueva manera de leer dos proteínas a la vez
La mayoría de los modelos de lenguaje proteico actuales tratan cada cadena proteica como si estuviera sola, ignorando cómo podría interactuar con sus parejas. Los autores, en cambio, construyeron un “Modelo de Lenguaje de Pares de Proteínas” (PPLM) que siempre observa dos secuencias una al lado de la otra. Emplea una arquitectura transformer, un tipo de modelo de aprendizaje profundo popular en tecnologías del lenguaje, pero personalizada para poder seguir por separado los patrones dentro de cada proteína y entre las dos proteínas. Para entrenarlo, el equipo recopiló más de 3,3 millones de pares de proteínas a partir de bases de datos estructurales y redes de interacción, dando al modelo una visión amplia de cómo tienden a emparejarse las proteínas naturales en la biología real.
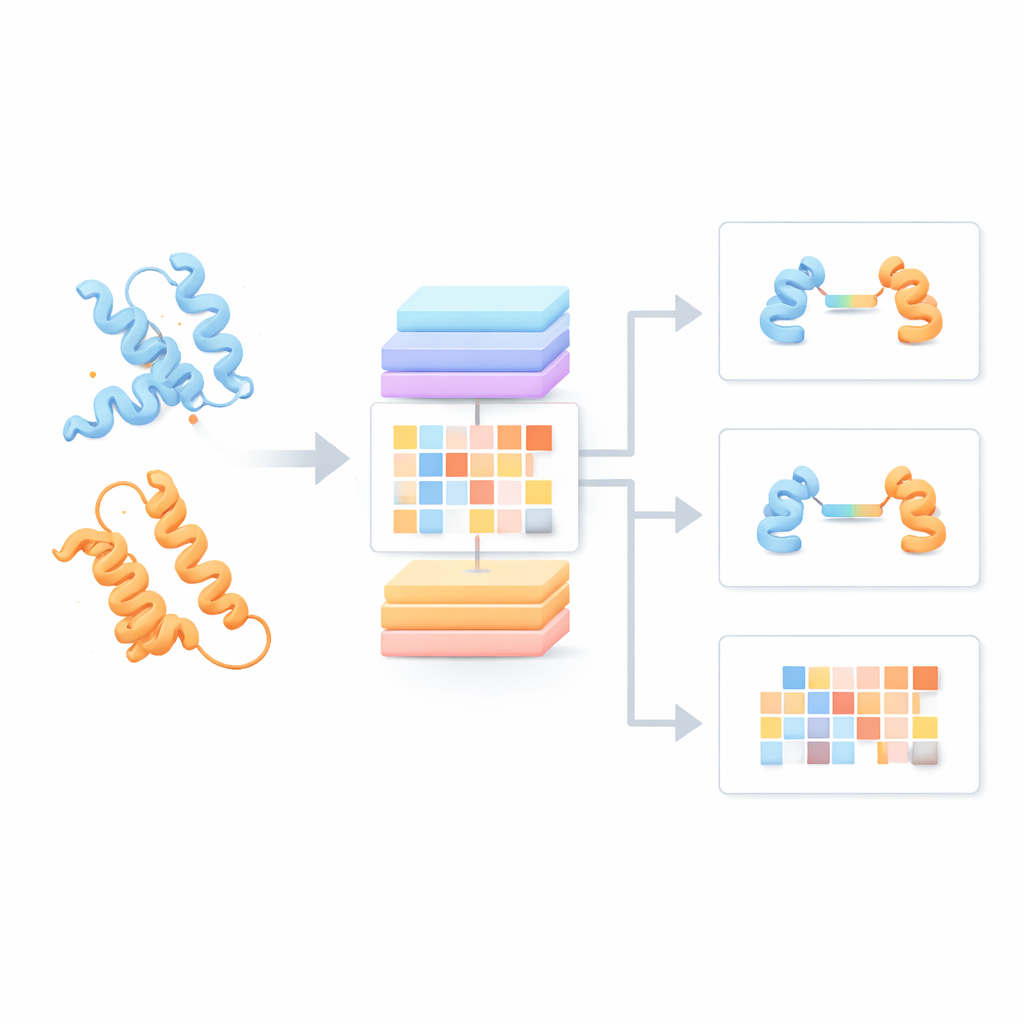
Detectando señales de interacción ocultas en las secuencias
Para comprobar si PPLM realmente comprende las asociaciones proteicas, los autores le pidieron que predijera aminoácidos enmascarados en pares de secuencias y lo compararon con un modelo líder de secuencia única llamado ESM2. En miles de pares de proteínas procedentes de distintas fuentes, PPLM fue consistentemente más confiado y más preciso, especialmente en residuos situados justo en la interfaz donde las proteínas se tocan. Al visualizar los patrones internos de “atención” del modelo, los investigadores mostraron que PPLM se centra de forma natural en esas regiones de contacto, aun cuando nunca se le indicó explícitamente dónde está la interfaz. En un estudio de caso detallado de un complejo proteico conocido, los pares de residuos con mayor atención del modelo coincidieron con la mayoría de los contactos observados experimentalmente en el espacio tridimensional.
De la comprensión básica a predicciones prácticas
Sobre esta base, el equipo creó tres herramientas aplicadas. PPLM-PPI predice si es probable que dos proteínas interactúen en absoluto. Probado en cinco especies diferentes, superó a varios métodos de vanguardia basados en secuencias, ofreciendo llamadas de interacción más precisas y más estables incluso cuando las proteínas de prueba eran bastante distintas de las vistas en el entrenamiento. PPLM-Affinity estima cuán fuerte es la unión entre dos proteínas. En una gran referencia de complejos con afinidades medidas, no solo superó a una versión de ESM2 afinada para la misma tarea, sino que también aventajó a un método especializado que usa estructuras 3D detalladas. Las mejoras fueron especialmente notables en sistemas de interés médico, como anticuerpos uniéndose a antígenos y receptores T reconociendo blancos inmunes.
Localizando con precisión dónde se tocan las proteínas
La tercera herramienta, PPLM-Contact, se centra en qué pares de residuos entre dos proteínas hacen contacto real. Combina los patrones de atención cruzada de PPLM con información evolutiva de alineamientos múltiples de secuencias y mapas de distancias de estructuras proteicas individuales. En varios conjuntos de prueba desafiantes, PPLM-Contact recuperó con precisión mapas de contacto e identificó residuos de interfaz mejor que los métodos existentes, incluidos algunos que dependen en gran medida de insumos estructurales. Una versión mejorada, PPLM-Contact2, va un paso más allá incorporando estructuras de complejos predichas por sistemas modernos de modelado 3D. Este enfoque híbrido mejora la predicción de contactos incluso más allá de esos predictores de estructura, ofreciendo vistas más nítidas de las superficies de unión y una localización más fiable de los sitios de unión.
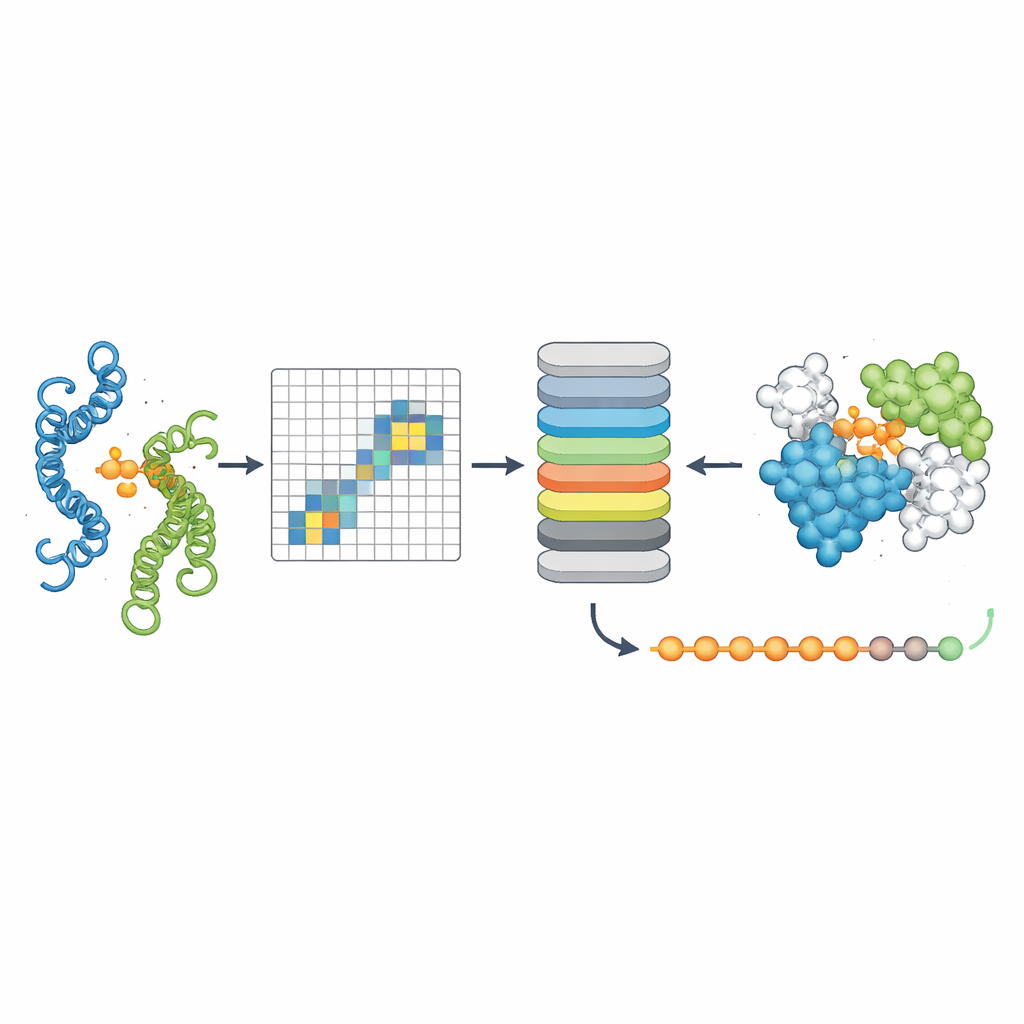
Qué supone esto para la biología y la medicina
En conjunto, este trabajo demuestra que leer secuencias de proteínas en pares, en lugar de aisladamente, permite a los modelos de IA captar patrones sutiles de interacción que sustentan la vida celular. PPLM y sus derivados pueden decir si dos proteínas probablemente se encontrarán, con qué fuerza se abrazarán y qué aminoácidos conforman el apretón —todo a partir de información de secuencia que es económica y abundante. Aunque el enfoque aún presenta dificultades con interfaces muy pequeñas o débiles y depende de la diversidad de los datos de entrenamiento disponibles, ofrece una vía escalable para mapear redes de interacción y guiar el diseño de anticuerpos, receptores T y otros fármacos biológicos. En esencia, el estudio demuestra que los modelos de lenguaje co-representados pueden transformar datos de secuencia crudos en conocimientos ricos y conscientes de la interacción sobre cómo las proteínas trabajan juntas.
Cita: Liu, J., Chen, H. & Zhang, Y. A paired sequence language model for protein-protein interaction modeling. Nat Commun 17, 3733 (2026). https://doi.org/10.1038/s41467-026-70457-5
Palabras clave: interacciones proteína–proteína, modelos de lenguaje de proteínas, afinidad de unión, predicción de contactos en la interfaz, biología estructural computacional