Clear Sky Science · de
Ein gepaartes Sequenz-Sprachmodell zur Modellierung von Protein-Protein-Interaktionen
Warum das Studium von Proteinpartnerschaften wichtig ist
Innerhalb jeder Zelle arbeiten Proteine selten allein. Sie bilden Paare oder größere Verbünde, um Signale weiterzugeben, Strukturen aufzubauen und Infektionen abzuwehren. Zu wissen, welche Proteine miteinander interagieren, wie fest sie binden und an welchen Stellen sie genau Kontakt haben, kann aufdecken, wie Zellen funktionieren und wie Krankheiten entstehen. Alle diese Proteinpartnerschaften im Labor zu messen, ist jedoch langsam und teuer. Diese Studie stellt ein neues Modell der künstlichen Intelligenz vor, das Proteinsequenzen paarweise einliest und lernt zu erkennen, wer wen bindet, wie stark und an welchen Kontaktpunkten — allein auf Basis der Aminosäuresequenzen als Eingabe.
Eine neue Art, zwei Proteine gleichzeitig zu lesen
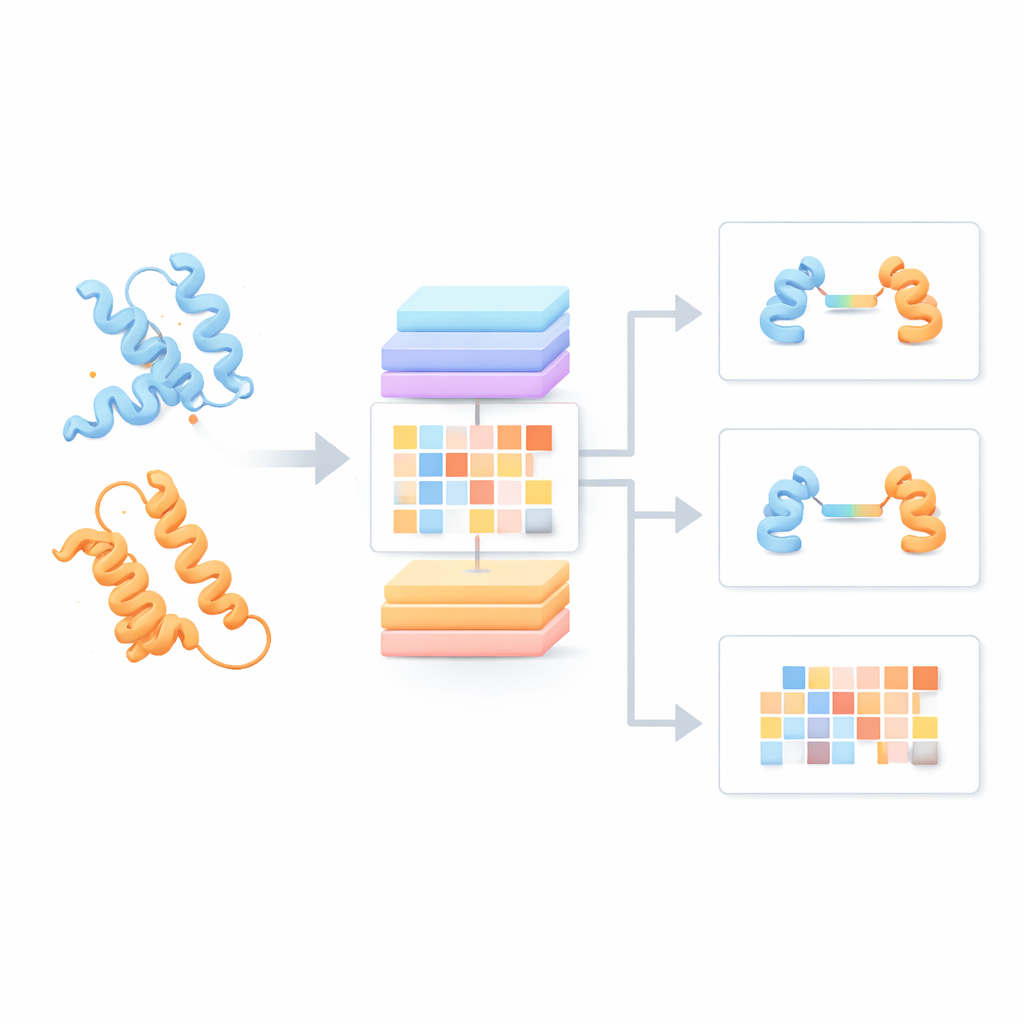
Die meisten aktuellen Protein-Sprachmodelle behandeln jede Proteinkette so, als stünde sie für sich, und ignorieren, wie sie mit Partnern interagieren könnte. Die Autoren entwickelten stattdessen ein "Protein Pair Language Model" (PPLM), das stets zwei Sequenzen nebeneinander betrachtet. Es verwendet eine Transformer-Architektur, eine in Sprachtechnologien verbreitete Form tiefen Lernens, wurde aber so angepasst, dass es Muster innerhalb jedes Proteins und zwischen den beiden Proteinen separat nachverfolgen kann. Zum Trainieren sammelte das Team über 3,3 Millionen Proteinpaare aus Strukturdatensätzen und Interaktionsnetzwerken, wodurch das Modell einen breiten Überblick darüber erhält, wie sich natürliche Proteine in der Biologie paaren.
Interaktionssignale, verborgen in Sequenzen, sichtbar machen
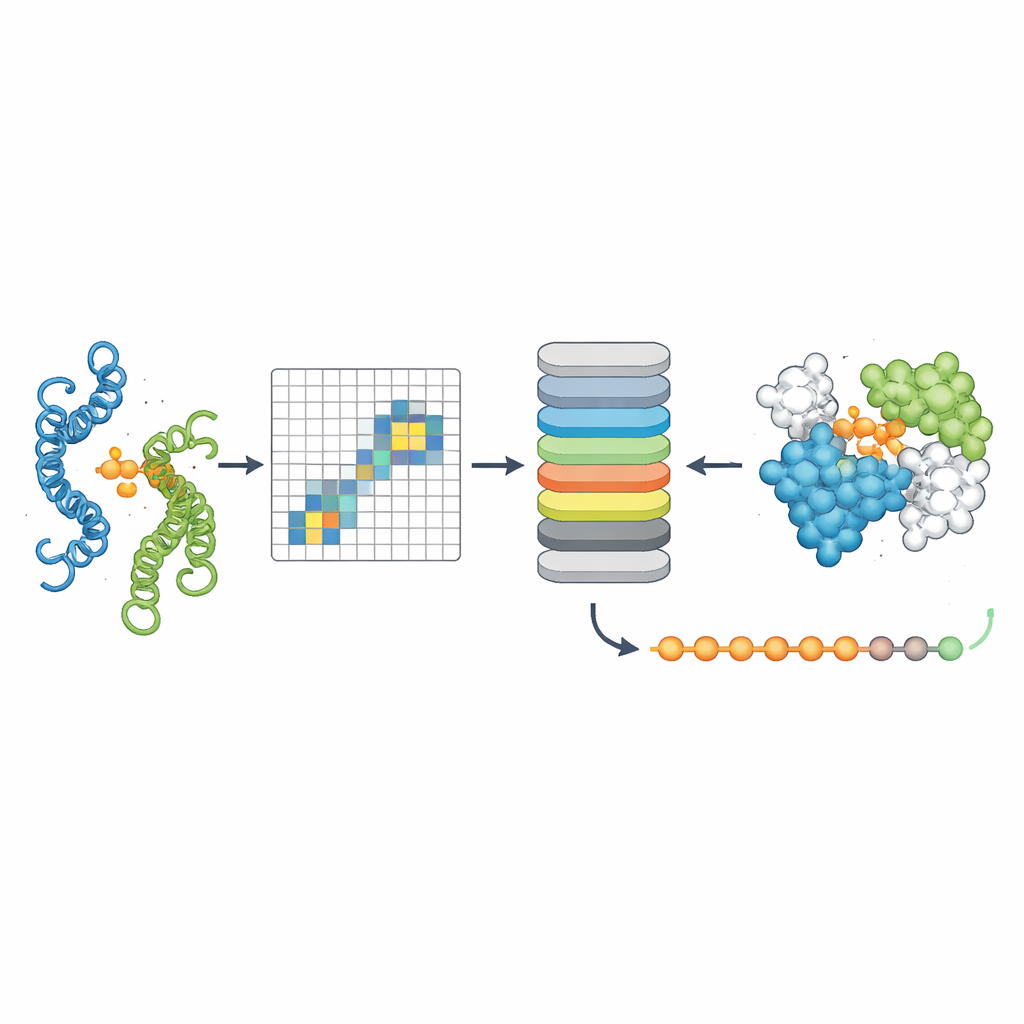
Um zu prüfen, ob PPLM tatsächlich Proteinpartnerschaften versteht, ließen die Autoren es maskierte Aminosäuren in Sequenzpaaren vorhersagen und verglichen die Ergebnisse mit einem führenden Ein-Seq-Modell namens ESM2. Über Tausende von Proteinpaaren aus verschiedenen Quellen war PPLM konsequent selbstsicherer und genauer, besonders bei Resten, die sich direkt an der Schnittstelle befinden, wo die Proteine Kontakt haben. Durch die Visualisierung der internen "Attention"-Muster des Modells zeigten die Forschenden, dass sich PPLM natürlicherweise auf diese Kontaktregionen fokussiert, obwohl es nie explizit gesagt bekam, wo die Schnittstelle liegt. In einer detaillierten Fallstudie eines bekannten Proteinkomplexes stimmten die vom Modell am stärksten beachteten Restpaarungen mit den experimentell beobachteten Kontakten im dreidimensionalen Raum weitgehend überein.
Von grundlegendem Verständnis zu praktischen Vorhersagen
Auf dieser Grundlage baute das Team drei angewandte Werkzeuge. PPLM-PPI sagt vorher, ob zwei Proteine wahrscheinlich überhaupt miteinander interagieren. Getestet über fünf verschiedene Arten hinweg übertraf es mehrere hochmoderne sequenzbasierte Methoden und lieferte genauere und stabilere Interaktionsvorhersagen, selbst wenn die Testproteine sich stark von denen im Training unterschieden. PPLM-Affinity schätzt, wie stark die Bindung zwischen zwei Proteinen ist. In einem großen Benchmark mit gemessenen Bindungsstärken von Komplexen übertraf es nicht nur eine für dieselbe Aufgabe feinabgestimmte Version von ESM2, sondern auch eine spezialisierte Methode, die detaillierte 3D-Strukturen nutzt. Die Verbesserungen waren besonders auffällig bei medizinisch wichtigen Systemen wie Antikörper-Antigen-Bindungen und T‑Zell-Rezeptoren, die Immunziele erkennen.
Genau lokalisieren, wo Proteine sich berühren
Das dritte Werkzeug, PPLM-Contact, zoomt darauf, welche Restpaare zwischen zwei Proteinen tatsächlich Kontakt machen. Es kombiniert PPLMs cross-protein Attention-Muster mit evolutionären Informationen aus mehrfachen Sequenzalignments und Distanzkarten aus individuellen Proteinstrukturen. In mehreren anspruchsvollen Testsets stellte PPLM-Contact Kontaktkarten akkurat wieder her und identifizierte Schnittstellenreste besser als bestehende Methoden, einschließlich einiger, die stark auf strukturelle Eingaben angewiesen sind. Eine erweiterte Version, PPLM-Contact2, geht einen Schritt weiter, indem sie vorausgesagte Komplexstrukturen moderner 3D-Modellierungssysteme einbezieht. Dieser hybride Ansatz verbessert die Kontaktvorhersage sogar gegenüber den Strukturvorhersagern selbst und bietet schärfere Einblicke in Bindungsflächen sowie zuverlässigere Lokalisierung von Bindungsstellen.
Was das für Biologie und Medizin bedeutet
Zusammengefasst zeigt diese Arbeit, dass das Einlesen von Proteinsequenzen paarweise statt einzeln es KI-Modellen ermöglicht, subtile Interaktionsmuster zu erfassen, die dem zellulären Leben zugrunde liegen. PPLM und seine Ableitungen können sagen, ob zwei Proteine sich wahrscheinlich begegnen, wie fest sie sich halten und welche Aminosäuren den "Handschlag" bilden — und das allein aus Sequenzinformationen, die günstig und reichlich vorhanden sind. Obwohl der Ansatz bei sehr kleinen oder schwachen Schnittstellen noch Schwierigkeiten hat und von der Vielfalt der verfügbaren Trainingsdaten abhängt, bietet er einen skalierbaren Weg, Interaktionsnetzwerke zu kartieren und das Design von Antikörpern, T‑Zell‑Rezeptoren und anderen biologischen Therapeutika zu leiten. Im Kern zeigt die Studie, dass gemeinsam repräsentierte Sprachmodelle rohe Sequenzdaten in reichhaltige, interaktionsbewusste Einsichten darüber verwandeln können, wie Proteine zusammenarbeiten.
Zitation: Liu, J., Chen, H. & Zhang, Y. A paired sequence language model for protein-protein interaction modeling. Nat Commun 17, 3733 (2026). https://doi.org/10.1038/s41467-026-70457-5
Schlüsselwörter: Protein–Protein-Interaktionen, Protein-Sprachmodelle, Bindungsaffinität, Vorhersage von Schnittstellenkontakten, computationale Strukturbiologie