Clear Sky Science · fr
Un modèle de langage de séquences appariées pour la modélisation des interactions protéine–protéine
Pourquoi étudier les partenariats protéiques est important
À l’intérieur de chaque cellule, les protéines travaillent rarement seules. Elles s’associent par paires ou en complexes plus larges pour transmettre des signaux, construire des structures et combattre les infections. Savoir quelles protéines interagissent, avec quelle affinité elles se lient et précisément où elles se touchent peut révéler le fonctionnement cellulaire et l’origine des maladies. Mais mesurer expérimentalement l’ensemble de ces partenariats protéiques est lent et coûteux. Cette étude présente un nouveau modèle d’intelligence artificielle qui lit des paires de séquences protéiques simultanément, apprenant à reconnaître qui se lie à qui, à quelle intensité et en quels points de contact — en n’utilisant que leurs séquences d’acides aminés comme entrée.
Une nouvelle manière de lire deux protéines à la fois
La plupart des modèles de langage pour protéines actuels traitent chaque chaîne protéique comme si elle était isolée, en ignorant les possibilités d’interaction avec des partenaires. Les auteurs ont construit à la place un « modèle de langage de paires de protéines » (PPLM) qui considère systématiquement deux séquences côte à côte. Il repose sur une architecture Transformer, un type de modèle d’apprentissage profond popularisé par les technologies du langage, mais adapté pour suivre séparément les motifs au sein de chaque protéine et entre les deux protéines. Pour l’entraîner, l’équipe a assemblé plus de 3,3 millions de paires de protéines issues de bases de données structurales et de réseaux d’interaction, offrant au modèle une vision large de la façon dont les protéines naturelles s’apparient en biologie réelle.
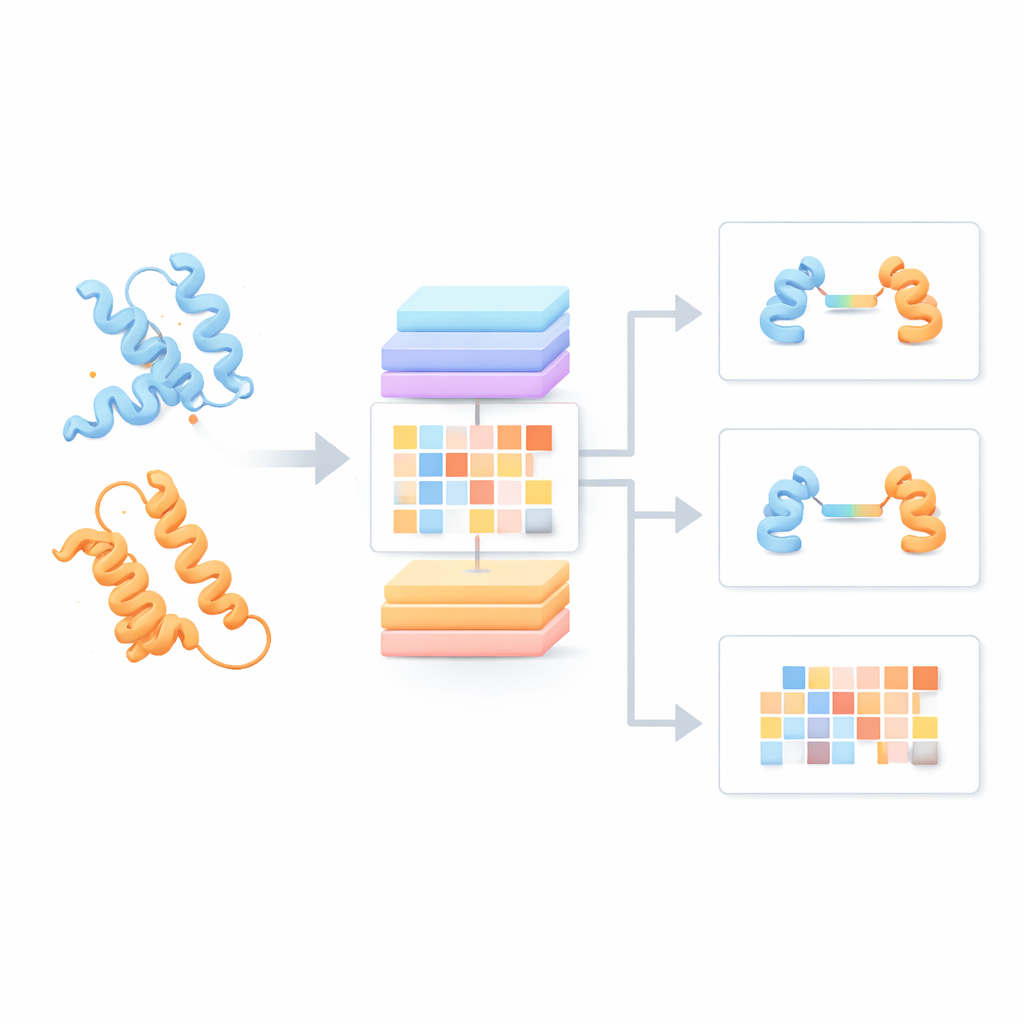
Dénicher des signaux d’interaction cachés dans les séquences
Pour évaluer si PPLM comprend véritablement les partenariats protéiques, les auteurs lui ont demandé de prédire des acides aminés masqués dans des paires de séquences et l’ont comparé à un modèle de séquence unique de référence, ESM2. Sur des milliers de paires protéiques issues de sources variées, PPLM s’est avéré systématiquement plus confiant et plus précis, en particulier aux résidus situés exactement à l’interface où les protéines se touchent. En visualisant les motifs d’« attention » internes du modèle, les chercheurs ont montré que PPLM se concentre naturellement sur ces régions de contact, bien qu’on ne lui ait jamais explicitement indiqué où se trouvent les interfaces. Dans une étude de cas détaillée d’un complexe protéique connu, les paires de résidus les plus fortement attendues par le modèle correspondaient à la plupart des contacts observés expérimentalement en trois dimensions.
De la compréhension fondamentale aux prédictions pratiques
Sur cette base, l’équipe a développé trois outils appliqués. PPLM-PPI prédit si deux protéines sont susceptibles d’interagir. Testé sur cinq espèces différentes, il a surpassé plusieurs méthodes séquentielles de pointe, fournissant des prédictions d’interaction plus précises et plus stables, même lorsque les protéines testées différaient fortement de celles vues pendant l’entraînement. PPLM-Affinity estime la force de liaison entre deux protéines. Sur un large banc d’essai de complexes avec mesures d’affinité, il a non seulement devancé une version d’ESM2 adaptée à la même tâche, mais aussi surpassé une méthode spécialisée qui utilise des structures 3D détaillées. Les améliorations ont été particulièrement marquées pour des systèmes d’importance médicale comme la liaison anticorps–antigènes et la reconnaissance par les récepteurs des cellules T de cibles immunitaires.
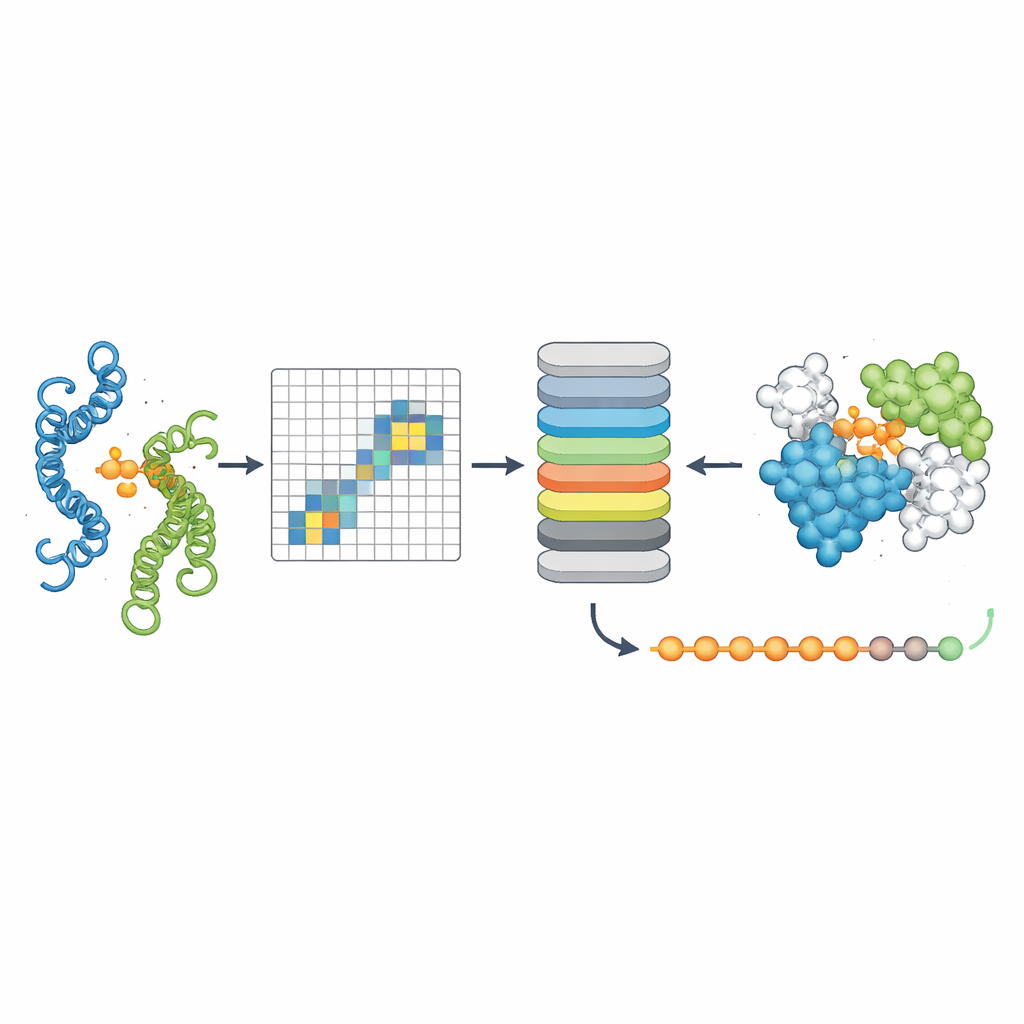
Localiser précisément où les protéines se touchent
Le troisième outil, PPLM-Contact, relève quelles paires de résidus entre deux protéines établissent effectivement un contact. Il combine les motifs d’attention inter-protéines de PPLM avec l’information évolutive provenant d’alignements de séquences multiples et des cartes de distances issues des structures individuelles. Sur plusieurs jeux de test difficiles, PPLM-Contact a reconstitué avec précision des cartes de contacts et identifié les résidus d’interface mieux que les méthodes existantes, y compris certaines fortement dépendantes d’entrées structurelles. Une version améliorée, PPLM-Contact2, va plus loin en intégrant des structures prédites de complexes par des systèmes modernes de modélisation 3D. Cette approche hybride améliore la prédiction des contacts au-delà des performances de ces prédicteurs de structure eux-mêmes, offrant des vues plus nettes des surfaces de liaison et une localisation plus fiable des sites d’interaction.
Ce que cela signifie pour la biologie et la médecine
En résumé, ce travail montre que lire les séquences protéiques par paires, plutôt qu’isolément, permet aux modèles d’IA de capturer des motifs d’interaction subtils qui sous-tendent la vie cellulaire. PPLM et ses dérivés peuvent indiquer si deux protéines sont susceptibles de se rencontrer, avec quelle force elles se lient et quels acides aminés constituent la « poignée de main » — tout cela à partir d’informations de séquence, bon marché et abondantes. Bien que l’approche ait encore des limites pour les interfaces très petites ou faibles et dépende de la diversité des données d’entraînement disponibles, elle offre une voie évolutive pour cartographier les réseaux d’interaction et guider la conception d’anticorps, de récepteurs de cellules T et d’autres médicaments biologiques. En substance, l’étude démontre que des modèles de langage co-représentés peuvent transformer des données de séquences brutes en informations riches et sensibles aux interactions sur la façon dont les protéines travaillent ensemble.
Citation: Liu, J., Chen, H. & Zhang, Y. A paired sequence language model for protein-protein interaction modeling. Nat Commun 17, 3733 (2026). https://doi.org/10.1038/s41467-026-70457-5
Mots-clés: interactions protéine–protéine, modèles de langage pour protéines, affinité de liaison, prédiction des contacts d’interface, biologie structurale computationnelle