Clear Sky Science · it
Un modello linguistico a sequenze accoppiate per la modellizzazione delle interazioni proteina-proteina
Perché studiare le partnership proteiche è importante
All’interno di ogni cellula, le proteine raramente agiscono da sole. Si associano in coppie o in gruppi più numerosi per trasmettere segnali, costruire strutture e contrastare infezioni. Sapere quali proteine interagiscono, con quale forza si legano e dove esattamente si toccano può rivelare come funzionano le cellule e come insorgono le malattie. Ma misurare in laboratorio tutte queste partnership proteiche è lento e costoso. Questo studio presenta un nuovo modello di intelligenza artificiale che legge coppie di sequenze proteiche insieme, imparando a riconoscere chi si lega a chi, quanto saldamente e in quali punti di contatto—usando come input solo le loro sequenze di amminoacidi.
Un nuovo modo di leggere due proteine contemporaneamente
La maggior parte degli attuali modelli linguistici per proteine tratta ogni catena proteica come se fosse isolata, ignorando come potrebbe interagire con partner. Gli autori hanno invece costruito un "Protein Pair Language Model" (PPLM) che considera sempre due sequenze affiancate. Utilizza un’architettura transformer, un tipo di modello di deep learning reso popolare dalle tecnologie linguistiche, ma personalizzata in modo da poter tracciare separatamente i pattern all'interno di ciascuna proteina e quelli tra le due proteine. Per addestrarlo, il gruppo ha raccolto oltre 3,3 milioni di coppie proteiche da database strutturali e reti di interazione, offrendo al modello una visione ampia di come le proteine naturali tendono ad associarsi nella biologia reale.
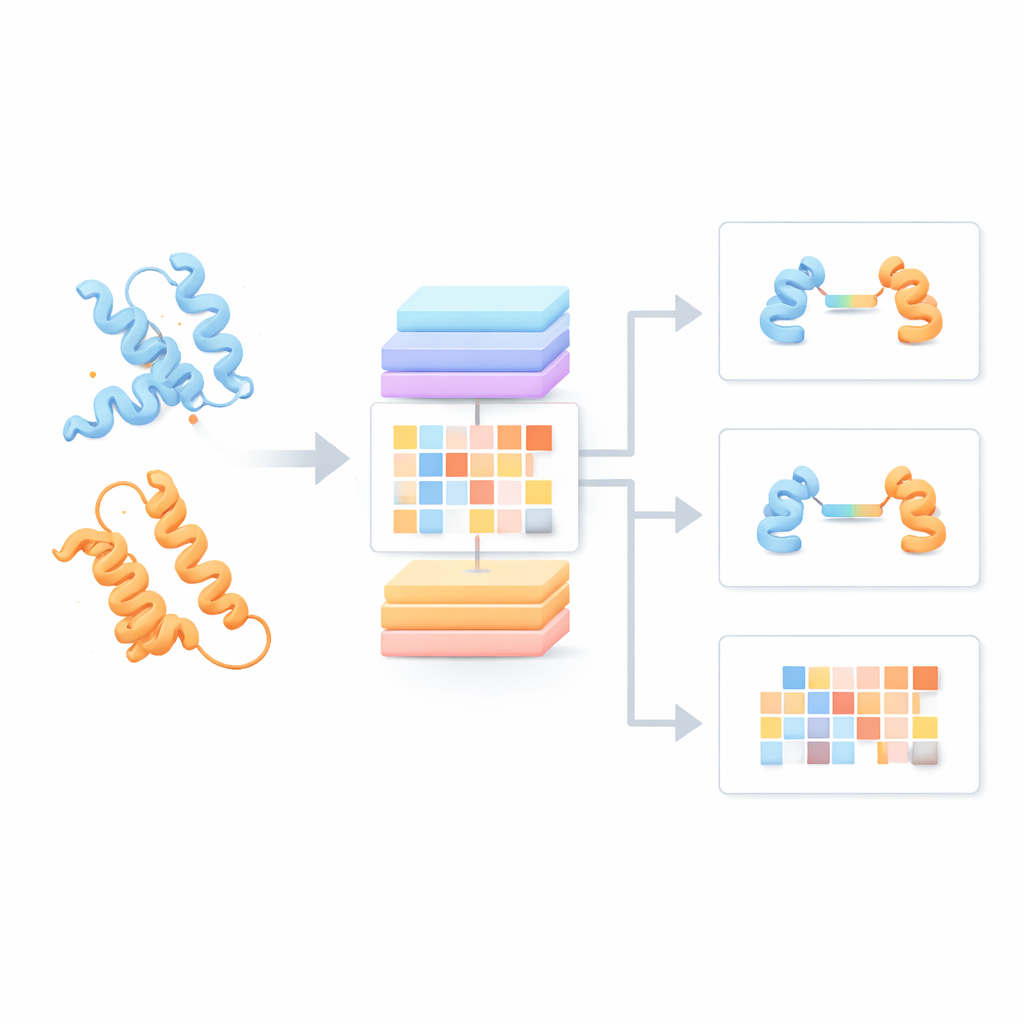
Scoprire segnali di interazione nascosti nelle sequenze
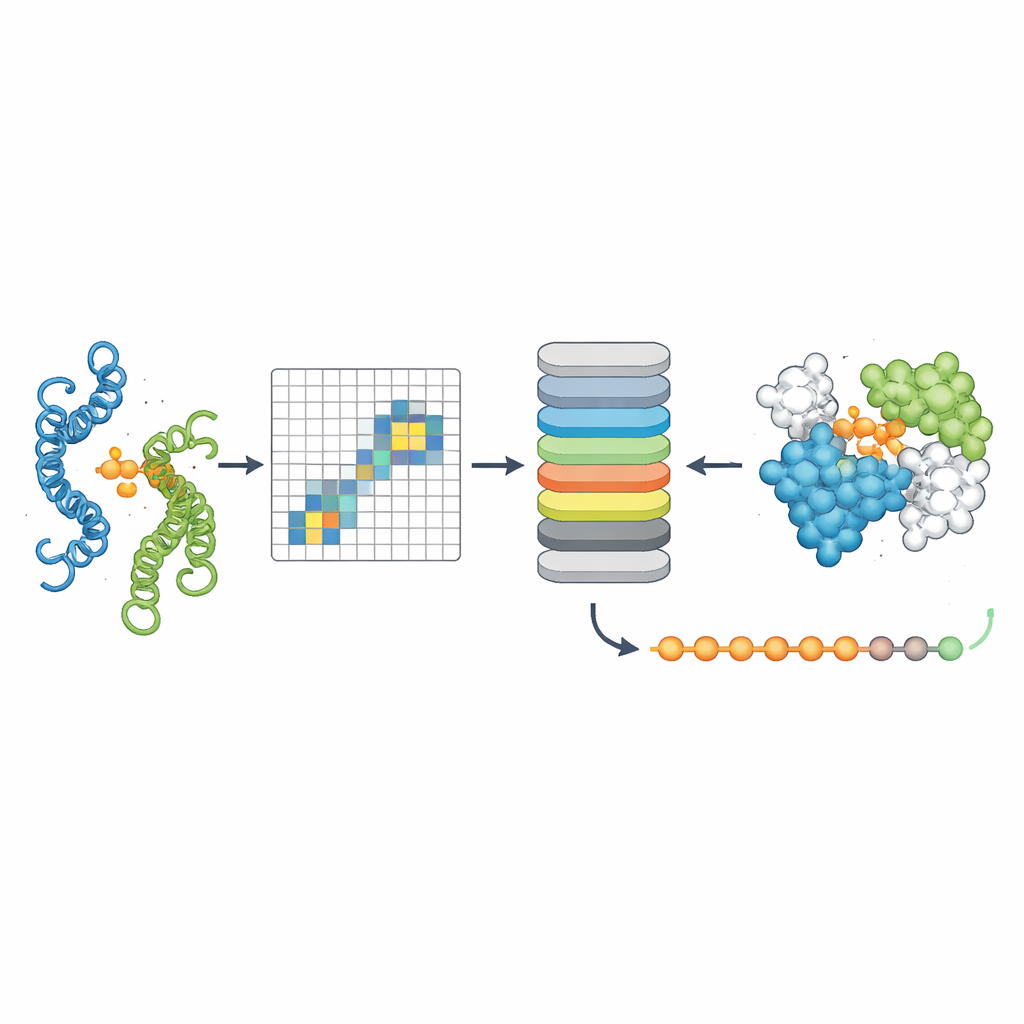
Per verificare se PPLM comprendesse davvero le partnership proteiche, gli autori gli hanno chiesto di predire amminoacidi mascherati in coppie di sequenze e lo hanno confrontato con un modello leader a singola sequenza chiamato ESM2. Su migliaia di coppie proteiche provenienti da diverse sorgenti, PPLM è risultato costantemente più sicuro e accurato, specie per i residui situati proprio all’interfaccia dove le proteine si toccano. Visualizzando i pattern di "attenzione" interni del modello, i ricercatori hanno mostrato che PPLM si focalizza naturalmente su quelle regioni di contatto, anche se non gli è mai stato detto esplicitamente dove si trovi l’interfaccia. In uno studio di caso dettagliato su un complesso proteico noto, le coppie di residui a cui il modello assegnava maggiore attenzione corrispondevano alla maggior parte dei contatti osservati sperimentalmente nello spazio tridimensionale.
Dalla comprensione di base alle predizioni pratiche
Sulla base di questo nucleo, il team ha creato tre strumenti applicativi. PPLM-PPI predice se è probabile che due proteine interagiscano. Testato su cinque diverse specie, ha superato diversi metodi basati sulle sequenze all’avanguardia, fornendo chiamate di interazione più accurate e più stabili anche quando le proteine di test erano abbastanza diverse da quelle viste in addestramento. PPLM-Affinity stima quanto è forte il legame tra due proteine. Su un ampio benchmark di complessi con forze di legame misurate, non solo ha battuto una versione di ESM2 rifinita per lo stesso compito, ma ha anche superato un metodo specializzato che utilizza strutture 3D dettagliate. I guadagni sono stati particolarmente evidenti per sistemi di rilevanza medica, come anticorpi che si legano ad antigeni e recettori T che riconoscono bersagli immunitari.
Individuare precisamente dove le proteine si toccano
Il terzo strumento, PPLM-Contact, si concentra su quali coppie di residui tra due proteine effettivamente entrano in contatto. Combina i pattern di attenzione cross-proteina di PPLM con informazioni evolutive provenienti da allineamenti multipli di sequenze e mappe di distanza tratte da strutture proteiche individuali. Su diversi set di test difficili, PPLM-Contact ha ricostruito accuratamente le mappe di contatto e identificato meglio i residui d’interfaccia rispetto ai metodi esistenti, inclusi alcuni che si basano pesantemente su input strutturali. Una versione potenziata, PPLM-Contact2, compie un ulteriore passo integrando strutture di complessi predette dai moderni sistemi di modellizzazione 3D. Questo approccio ibrido migliora la predizione dei contatti anche rispetto a quei predittori di struttura, offrendo visioni più nitide delle superfici di legame e una localizzazione più affidabile dei siti di interazione.
Cosa significa per la biologia e la medicina
Nel complesso, questo lavoro mostra che leggere le sequenze proteiche a coppie, invece che singolarmente, permette ai modelli di intelligenza artificiale di catturare pattern sottili di interazione che sottendono la vita cellulare. PPLM e le sue derivate possono dire se due proteine è probabile che si incontrino, quanto strettamente si legano e quali amminoacidi costituiscono la "stretta di mano"—tutto a partire da informazioni di sequenza economiche e abbondanti. Sebbene l’approccio fatichi ancora con interfacce molto piccole o deboli e dipenda dalla diversità dei dati di addestramento disponibili, offre una via scalabile per mappare reti di interazione e guidare il disegno di anticorpi, recettori T e altri farmaci biologici. In sostanza, lo studio dimostra che modelli linguistici co-rappresentati possono trasformare dati di sequenza grezzi in approfondimenti ricchi e consapevoli delle interazioni su come le proteine lavorano insieme.
Citazione: Liu, J., Chen, H. & Zhang, Y. A paired sequence language model for protein-protein interaction modeling. Nat Commun 17, 3733 (2026). https://doi.org/10.1038/s41467-026-70457-5
Parole chiave: interazioni proteina–proteina, modelli linguistici per proteine, affinità di legame, predizione dei contatti sull'interfaccia, biologia strutturale computazionale