Clear Sky Science · sv
Att rikta in sig på Prolyl 3-hydroxylase 1 hämmar bukspottkörtelcancerprogression och makrofagers immunfunktion
Varför denna forskning är viktig
Bukspottkörtelcancer hör till de dödligaste cancerformerna, upptäcks ofta sent och är ökänd för att vara motståndskraftig mot behandling. Denna studie undersöker en dold molekylär "strömbrytare" som hjälper pankreastumörer att växa och undkomma immunsystemet, och visar hur att stänga av den strömbrytaren kan bromsa sjukdomen och få konventionell cytostatika att fungera bättre.
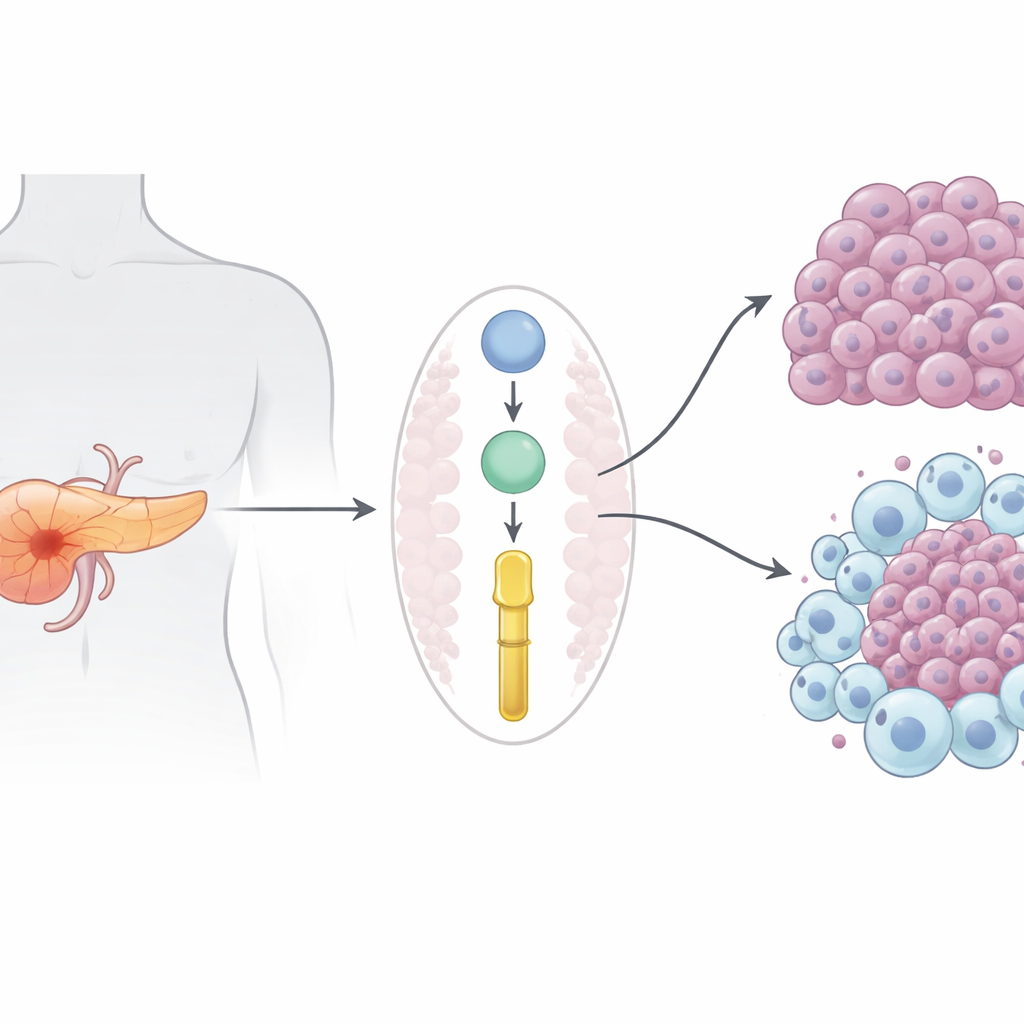
En dödlig cancer med få bra alternativ
Pancreas ductal adenocarcinoma, den vanligaste formen av pankreascancer, dödar nästan lika många som den drabbar. Eftersom den vanligtvis diagnostiseras efter att den redan spridit sig är kirurgi ofta inte möjlig, och läkemedel som gemcitabin ger bara måttlig och kortvarig nytta. Moderna immunterapier som revolutionerat melanom och lungcancer har i stort sett misslyckats här, delvis eftersom pankreastumörer skapar en immunologiskt "kall" miljö som håller ute hjälpsamma immunceller och gynnar celler som skyddar cancern.
Den dolda hjälparen: ett kollagenenzym som gått fel
Forskarna fokuserade på proteiner som tumörceller utsöndrar och som kan påverka både cancerns tillväxt och den omgivande immunmiljön. Genom att omanalysere stora proteindataset från mänskliga patienter och en väletablerad musmodell identifierade de Prolyl 3-hydroxylase 1 (P3H1), ett enzym mest känt för att modifiera kollagen i ben och hud. Hos både människor och möss var P3H1-nivåerna mycket högre i pankreastumörer än i frisk bukspottkörtel, och patienter vars tumörer hade mer P3H1 tenderade att recidivera tidigare och avlida snabbare. Viktigt är att ökat P3H1 kopplades till mer omfattande sjukdom, vilket tyder på att det gör mer än att bara följa tumörtillväxten.
Att stänga av P3H1 bromsar tumörer och omformar immunceller
För att testa om P3H1 faktiskt driver cancer konstruerade teamet möss där deras pankreasceller saknade P3H1-genen, samtidigt som de fortfarande bar de klassiska cancerframkallande mutationerna som normalt ger aggressiva tumörer. Dessa P3H1-defekta möss utvecklade mindre tumörer, behöll mer normalt pankreasvävnad och levde cirka 20 % längre än motsvarigheterna med intakt P3H1. I både mustumörer och mänskliga cancercellinjer odlade i labbet minskade tumörcellsproliferation kraftigt vid förlust av P3H1. Samtidigt innehöll tumörer som saknade P3H1 betydligt färre "M2-liknande" tumörassocierade makrofager — immunceller som vanligtvis hjälper tumörer genom att främja tillväxt, kärlbildning och läkemedelsresistens. Cancerceller med högt P3H1 utsöndrade mer av tre signalproteiner (CXCL1, CXCL5 och CXCL8) som lockar och omprogrammerar makrofager; att sänka P3H1 minskade dessa signaler och dämpade makrofagrekytering och polarisering.
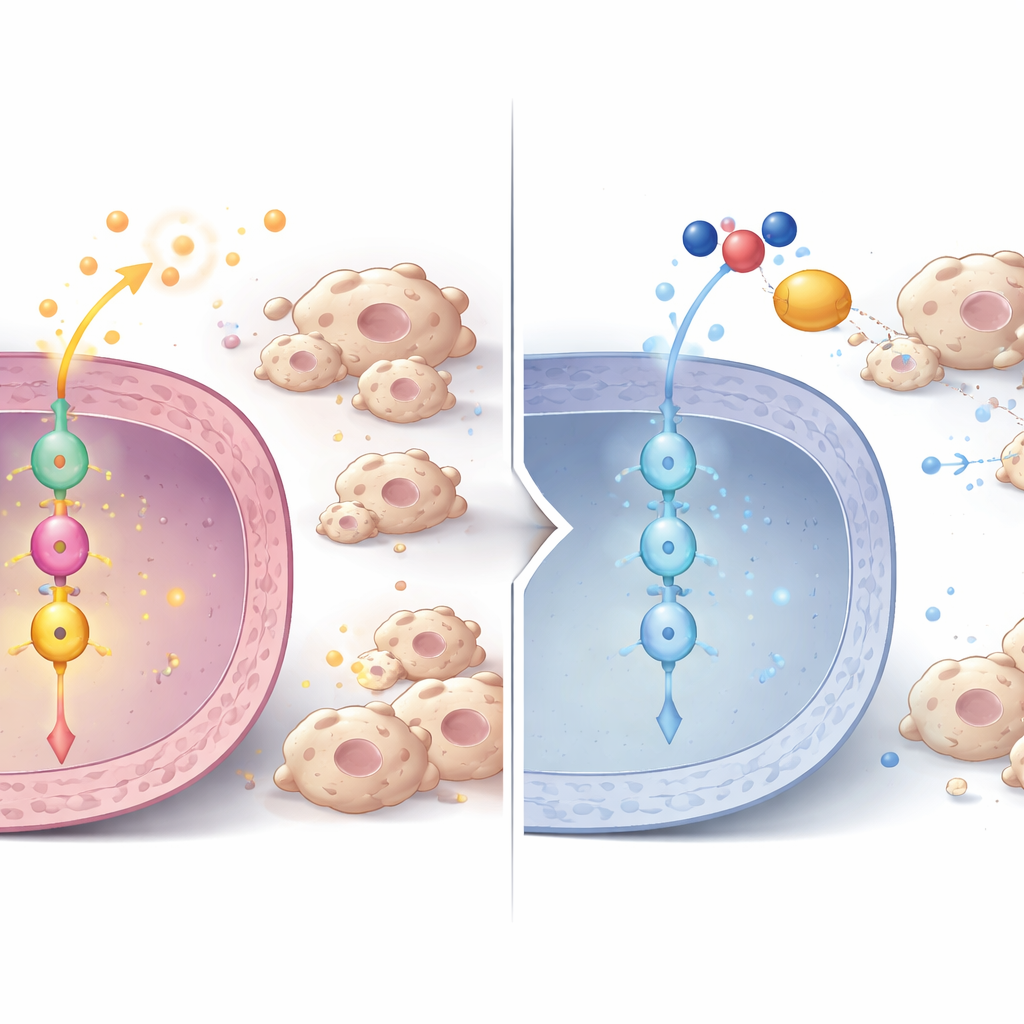
En signalväg från P3H1 till celldelning och immunkontroll
Vid djupare analys avslöjade forskarna en signalväg inne i tumörcellerna. När P3H1 minskades sjönk nivåerna av ett annat protein, Polo-like kinase 1 (PLK1), både på protein- och RNA-nivå, i celler, mustumörer och patientprover. PLK1 är en nyckelregulator av celldelning och är redan känd för att vara överaktiv i många cancerformer. Med hjälp av fosfoproteomik visade teamet att PLK1-aktivitet normalt förstärker en central cancerregulator kallad β-catenin genom att modifiera den vid en specifik plats som krävs för inträde i cellkärnan. I P3H1-defekta celler föll denna aktiverande markering på β-catenin, dess närvaro i kärnan minskade och uttrycket av β-catenintargetgener sjönk. Dessa mål inkluderar inte bara cellcykeldrivare som c-Myc och Cyclin D1, som driver celler att dela sig, utan också de cytokiner som lockar tumörstödjande makrofager. Återställande av antingen P3H1 eller PLK1 i P3H1-defekta celler återupplivade β-catenin-aktivitet, cancercellsproliferation och makrofagrekytering, vilket understryker att P3H1–PLK1–β-catenin-axeln är ett centralt kontrollnav.
Att göra cytostatika mer effektiv
Där β-catenin-signalering och tumörstödjande makrofager båda implicerats i resistens mot gemcitabin och andra standardregimer, frågade författarna om blockering av denna axel kunde förbättra behandlingen. De kombinerade gemcitabin (och andra standardcocktails) med en PLK1-hämmare, BI2536, i möss med mänskliga pankreastumörer. Vid en noggrant vald dos som undvek organtoxicitet gav kombinationen mycket mindre tumörer, färre delande cancerceller och färre tumörassocierade makrofager än cytostatika ensam. Patientderiverade pankreasorganoider — mini‑tumörer odlade från kirurgiska prover — visade ett liknande mönster: nedreglering av P3H1 bromsade deras tillväxt, sänkte PLK1 och gjorde dem mer sårbara för gemcitabin plus nab‑paklitaxel, medan PLK1‑hämmaren ytterligare ökade läkemedelskänsligheten.
Vad detta innebär för framtida behandling
Enkelt uttryckt identifierar detta arbete en trio molekyler — P3H1, PLK1 och β-catenin — som tillsammans fungerar som en gaspedal för pankreastumörer, driver både okontrollerad celldelning och rekrytering av immunceller som hjälper cancern istället för att bekämpa den. Att inaktivera denna axel bromsar tumörtillväxt och gör befintlig cytostatika mer effektiv i möss och patientderiverade modeller. Medan läkemedel som blockerar P3H1 direkt ännu inte finns, är PLK1‑hämmare redan under utveckling, vilket väcker möjligheten att kombinera dem med standardcytostatika i framtiden för att ge patienter med pankreascancer mer hållbar kontroll över sin sjukdom.
Citering: Bai, P., Liu, C., Fu, C. et al. Targeting Prolyl 3-hydroxylase 1 inhibits pancreatic cancer progression and macrophage immunity. Nat Commun 17, 3913 (2026). https://doi.org/10.1038/s41467-026-70452-w
Nyckelord: bukspottkörtelcancer, tumörmikromiljö, makrofager, β-catenin-signalering, målinriktad cytostatika