Clear Sky Science · pl
Celowanie w prolyl-3-hydroksylazę 1 hamuje postęp raka trzustki i wpływa na odporność makrofagów
Dlaczego te badania są ważne
Rak trzustki należy do najgroźniejszych nowotworów — często jest wykrywany późno i wykazuje znaczną oporność na leczenie. Badanie to bada ukryty molekularny „wyłącznik”, który sprzyja wzrostowi nowotworów trzustki i ich unikaniu przez układ odpornościowy, oraz pokazuje, że jego wyłączenie może spowolnić chorobę i poprawić skuteczność standardowej chemioterapii.
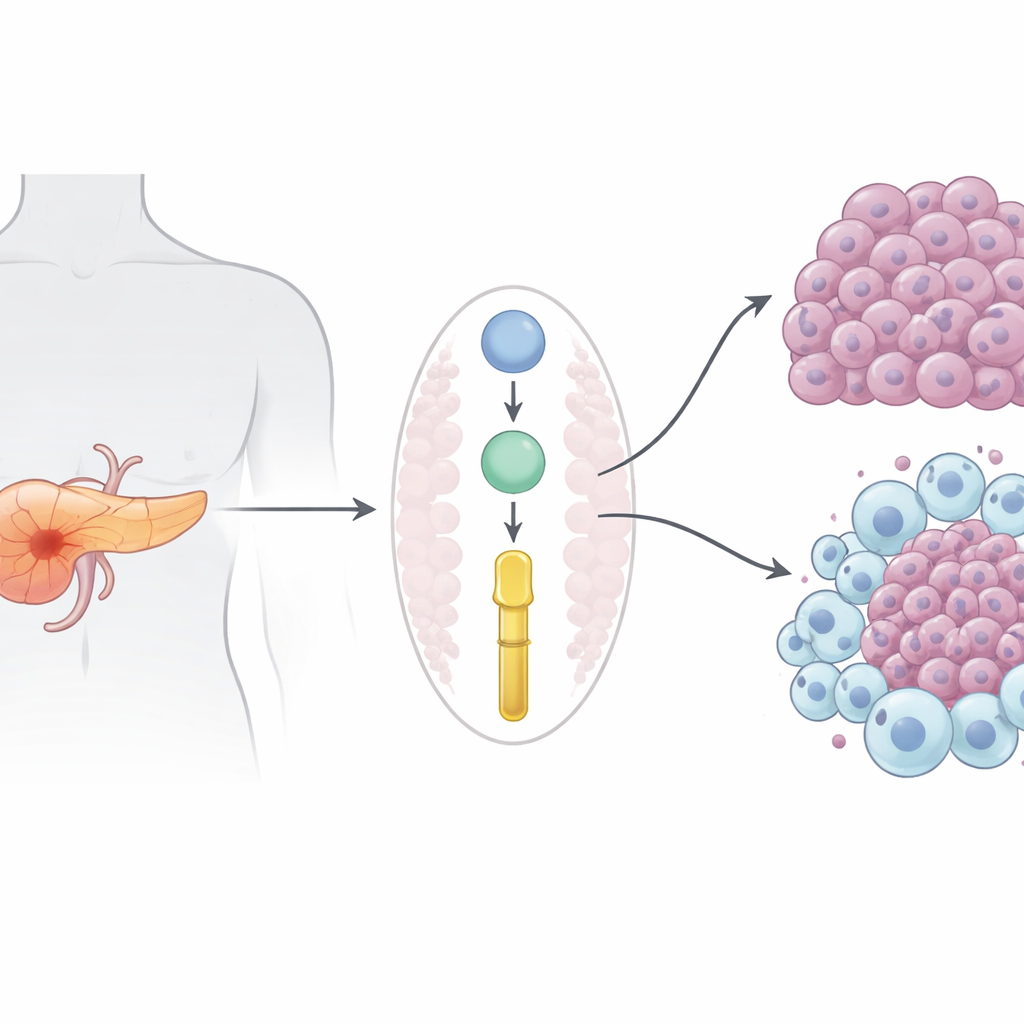
Śmiertelny nowotwór z niewieloma dobrymi opcjami
Gruczolakorak przewodowy trzustki, najczęstsza postać raka trzustki, zabija niemal tyle osób, ile diagnozuje się zachorowań. Ponieważ zwykle rozpoznawany jest dopiero po rozsianiu, często nie ma możliwości leczenia chirurgicznego, a leki takie jak gemcytabina przynoszą jedynie skromne i krótkotrwałe korzyści. Nowoczesne immunoterapie, które zrewolucjonizowały leczenie czerniaka i raka płuca, w przypadku raka trzustki zasadniczo zawiodły — częściowo dlatego, że guzy trzustki tworzą immunologicznie „zimne” środowisko, które odpycha pomocne komórki odpornościowe i sprzyja komórkom chroniącym nowotwór.
Ukryty wspomagacz: enzym kolagenowy na wariackich papierach
Naukowcy skupili się na białkach wydzielanych przez komórki nowotworowe, które mogą kształtować zarówno wzrost raka, jak i otaczające go środowisko immunologiczne. Przeanalizowawszy duże zbiory danych białkowych od pacjentów i w dobrze opisanym modelu mysim, wytypowali Prolyl 3-hydroxylase 1 (P3H1) — enzym znany głównie z modyfikacji kolagenu w kości i skórze. Zarówno u ludzi, jak i u myszy poziomy P3H1 były znacznie wyższe w guzach trzustki niż w zdrowej tkance trzustki, a pacjenci, których guzy wykazywały większe stężenie P3H1, mieli tendencję do szybszych nawrotów i krótszego przeżycia. Co istotne, zwiększone poziomy P3H1 wiązały się z bardziej zaawansowaną chorobą, sugerując, że P3H1 robi więcej niż tylko towarzyszy wzrostowi guza.
Wyłączenie P3H1 hamuje guzy i zmienia typ makrofagów
Aby sprawdzić, czy P3H1 rzeczywiście napędza raka, zespół stworzył myszy, których komórki trzustki nie zawierały genu P3H1, zachowując jednocześnie klasyczne mutacje powodujące agresywne guzy. Myszy pozbawione P3H1 rozwijały mniejsze guzy, zachowywały więcej prawidłowej tkanki trzustki i żyły około 20% dłużej niż ich odpowiedniki z niezmienionym P3H1. Zarówno w guzach mysich, jak i w hodowlach ludzkich linii komórkowych utrata P3H1 wyraźnie zmniejszała proliferację komórek nowotworowych. Równocześnie guzy pozbawione P3H1 zawierały znacznie mniej makrofagów „M2-podobnych” — komórek odpornościowych, które zwykle sprzyjają nowotworom, promując wzrost, tworzenie naczyń i oporność na leki. Komórki nowotworowe z wysokim P3H1 wydzielały więcej trzech białek sygnałowych (CXCL1, CXCL5 i CXCL8), które przyciągają i przeprogramowują makrofagi; obniżenie P3H1 zmniejszało te sygnały i ograniczało rekrutację oraz polaryzację makrofagów.
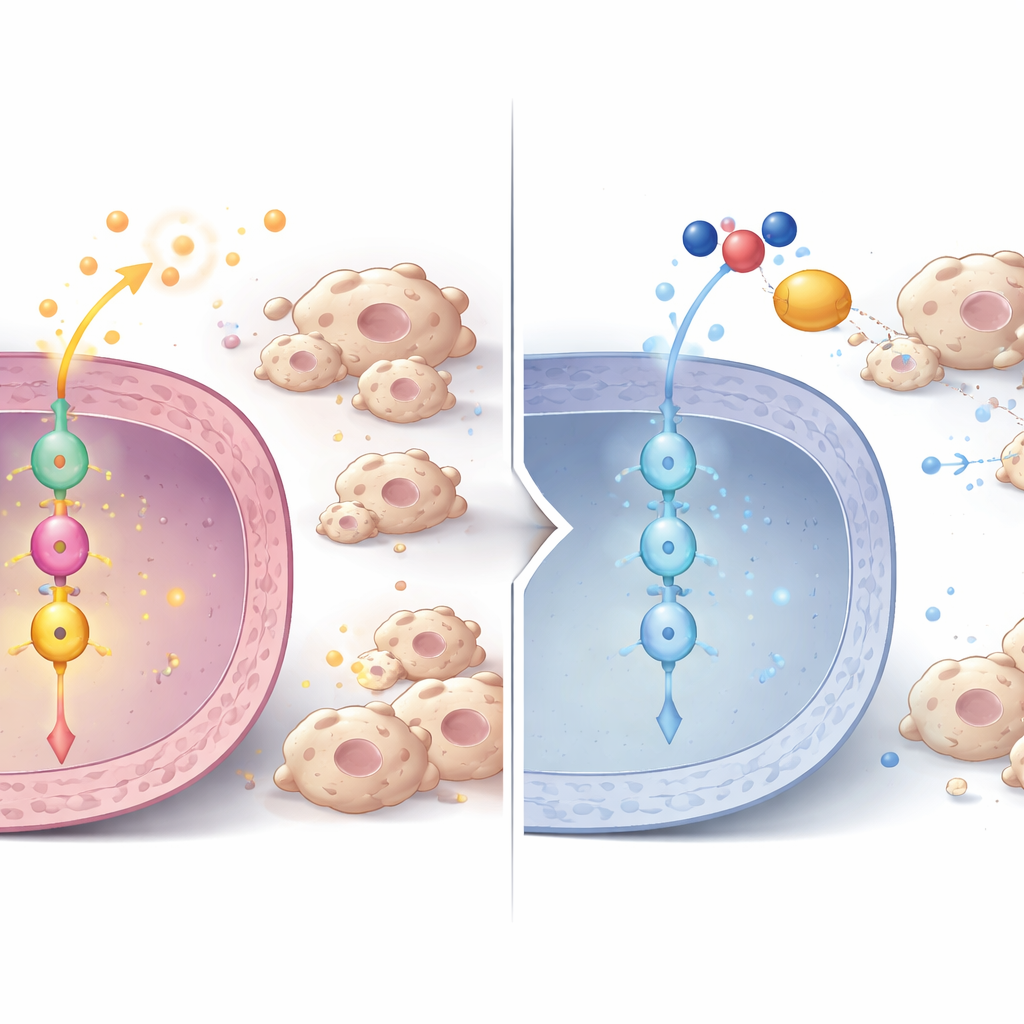
Łańcuch sygnałowy od P3H1 do podziału komórkowego i kontroli odporności
Badając mechanizm głębiej, naukowcy odkryli łańcuch sygnałowy wewnątrz komórek nowotworowych. Po zmniejszeniu P3H1 spadały poziomy innego białka — Polo-like kinazy 1 (PLK1) — zarówno na poziomie białka, jak i RNA, w komórkach, guzach mysich i próbkach pacjentów. PLK1 jest kluczowym regulatorem podziału komórkowego i wiadomo, że jest nadaktywna w wielu nowotworach. Dzięki fosfoproteomice zespół wykazał, że aktywność PLK1 zwykle wzmacnia centralnego regulatora nowotworowego, β-kateninę, poprzez modyfikację w określonym miejscu niezbędnym do wejścia do jądra komórkowego. W komórkach pozbawionych P3H1 spadła ta aktywująca modyfikacja β-kateniny, zmniejszyła się jej obecność w jądrze, a ekspresja genów docelowych β-kateniny obniżyła się. Do tych celów należą nie tylko promotory cyklu komórkowego, takie jak c-Myc i Cyclin D1, które pobudzają podziały, ale też cytokiny przyciągające makrofagi wspierające guz. Przywrócenie P3H1 lub PLK1 w komórkach pozbawionych P3H1 odnowiło aktywność β-kateniny, proliferację komórek nowotworowych i rekrutację makrofagów, co podkreśla znaczenie osi P3H1–PLK1–β-katenina jako centralnego węzła kontrolnego.
Wzmacnianie działania chemioterapii
Ponieważ sygnalizacja β-kateniny i makrofagi wspierające guz są powiązane z opornością na gemcytabinę i inne standardowe schematy, autorzy sprawdzili, czy blokowanie tej osi może poprawić leczenie. Połączyli gemcytabinę (i inne standardowe koktajle) z inhibitorem PLK1, BI2536, u myszy z implantowanymi ludzkimi guzami trzustki. Przy starannie dobranej dawce, unikającej toksyczności narządowej, kombinacja dała znacznie mniejsze guzy, mniej dzielących się komórek nowotworowych i mniej makrofagów związanych z guzem niż sama chemioterapia. Organoidy trzustkowe pochodzące od pacjentów — miniaturowe guzy hodowane z próbek chirurgicznych — wykazały podobny wzorzec: wyciszenie P3H1 spowalniało ich wzrost, obniżało PLK1 i zwiększało wrażliwość na gemcytabinę z nab-paklitakselem, a inhibitor PLK1 dodatkowo potęgował wrażliwość na leki.
Co to oznacza dla przyszłego leczenia
Mówiąc prosto, praca ta identyfikuje triadę cząsteczek — P3H1, PLK1 i β-kateninę — które działają jak pedał przyspieszenia dla guzów trzustki, napędzając zarówno niekontrolowane podziały komórkowe, jak i rekrutację komórek odpornościowych, które sprzyjają rakowi zamiast go zwalczać. Wyłączenie tej osi spowalnia wzrost guza i zwiększa skuteczność istniejącej chemioterapii w modelach mysich i pochodzących od pacjentów. Choć leki bezpośrednio blokujące P3H1 nie są jeszcze dostępne, inhibitory PLK1 są już w fazie rozwoju, co daje nadzieję, że w przyszłości ich połączenie ze standardową chemioterapią może zapewnić pacjentom z rakiem trzustki dłuższą kontrolę choroby.
Cytowanie: Bai, P., Liu, C., Fu, C. et al. Targeting Prolyl 3-hydroxylase 1 inhibits pancreatic cancer progression and macrophage immunity. Nat Commun 17, 3913 (2026). https://doi.org/10.1038/s41467-026-70452-w
Słowa kluczowe: rak trzustki, mikrośrodowisko guza, makrofagi, sygnalizacja β-kateniny, ukierunkowana chemioterapia