Clear Sky Science · en
Targeting Prolyl 3-hydroxylase 1 inhibits pancreatic cancer progression and macrophage immunity
Why this research matters
Pancreatic cancer is among the deadliest cancers, often detected late and notoriously resistant to treatment. This study explores a hidden molecular “switch” that helps pancreatic tumors grow and evade the immune system, and shows how turning that switch off can slow the disease and make standard chemotherapy work better.
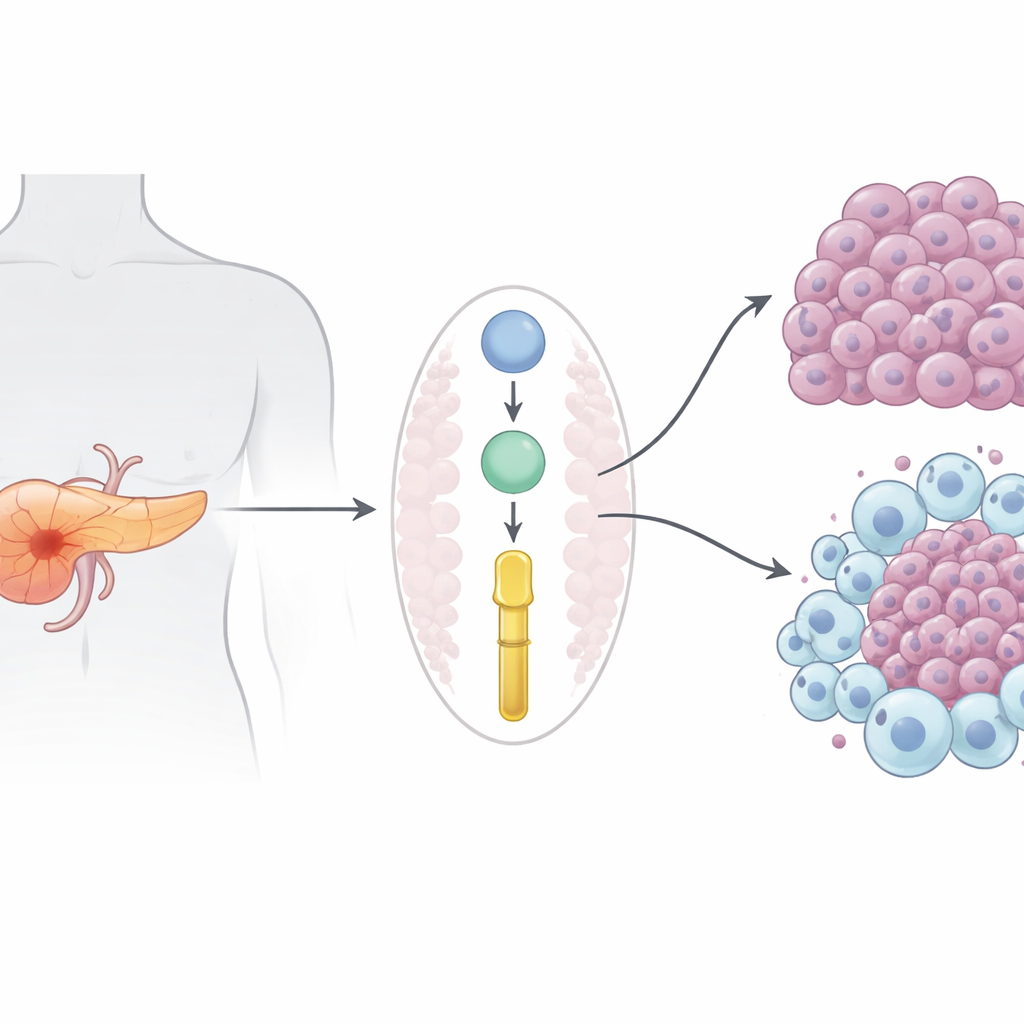
A deadly cancer with few good options
Pancreatic ductal adenocarcinoma, the most common form of pancreatic cancer, kills nearly as many people as it strikes. Because it is usually diagnosed after it has already spread, surgery is often not possible, and drugs such as gemcitabine give only modest and short-lived benefit. Modern immunotherapies that have transformed melanoma and lung cancer have largely failed here, in part because pancreatic tumors create an immunologically “cold” environment that keeps helpful immune cells out and nurtures cells that protect the cancer.
The hidden helper: a collagen enzyme gone rogue
The researchers focused on proteins secreted by tumor cells that might shape both cancer growth and the surrounding immune environment. By reanalyzing large protein datasets from human patients and a well-established mouse model, they pinpointed Prolyl 3-hydroxylase 1 (P3H1), an enzyme best known for modifying collagen in bone and skin. In both people and mice, P3H1 levels were far higher in pancreatic tumors than in healthy pancreas, and patients whose tumors had more P3H1 tended to relapse sooner and die earlier. Importantly, increased P3H1 was linked with more extensive disease, suggesting it does more than merely accompany tumor growth.
Turning P3H1 off slows tumors and reshapes immune cells
To test whether P3H1 is actually driving cancer, the team engineered mice so their pancreatic cells lacked the P3H1 gene, while still carrying the classic cancer-causing mutations that normally produce aggressive tumors. These P3H1-deficient mice developed smaller tumors, retained more normal pancreatic tissue, and lived about 20% longer than their counterparts with intact P3H1. In both mouse tumors and human cancer cell lines grown in the lab, loss of P3H1 sharply reduced tumor cell proliferation. At the same time, tumors missing P3H1 contained far fewer “M2-like” tumor-associated macrophages—immune cells that usually help tumors by promoting growth, blood vessel formation, and drug resistance. The cancer cells with high P3H1 secreted more of three signal proteins (CXCL1, CXCL5, and CXCL8) that attract and reprogram macrophages; dialing down P3H1 lowered these signals and blunted macrophage recruitment and polarization.
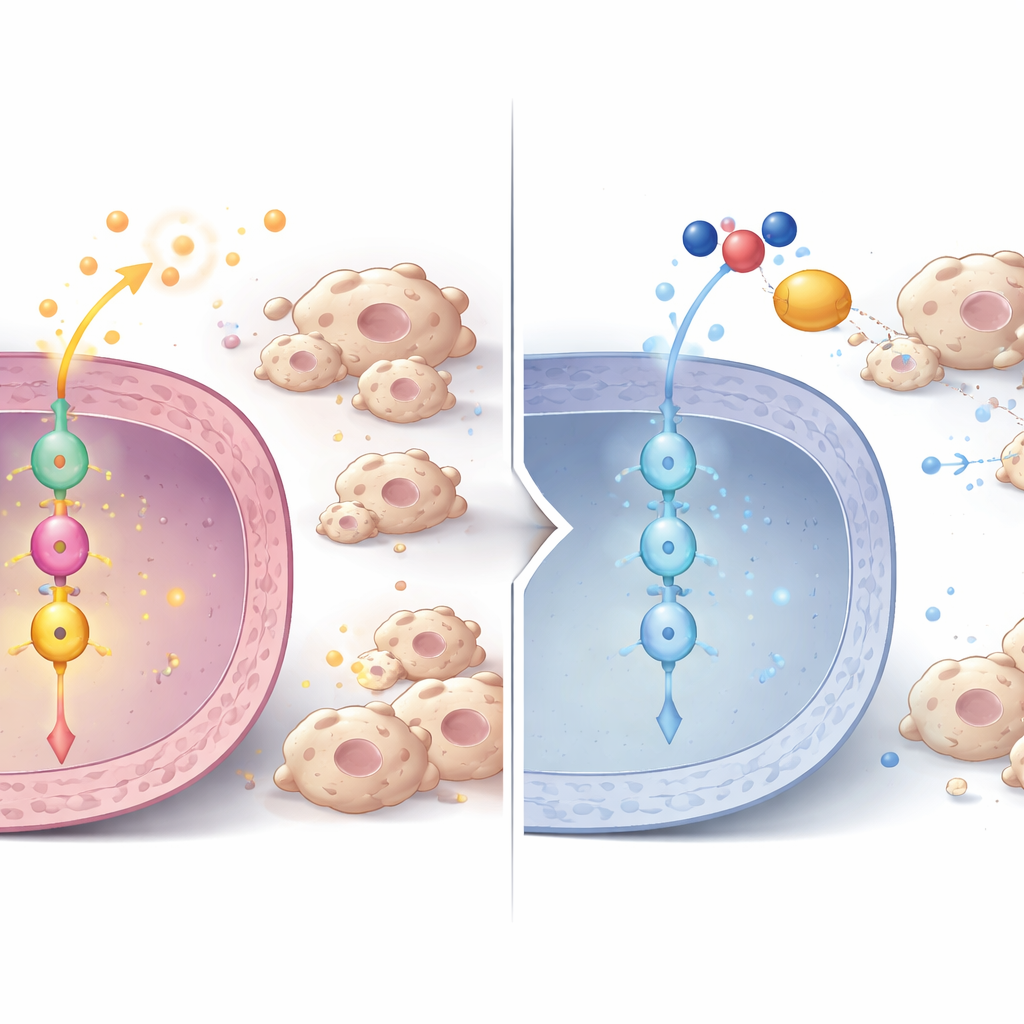
A signaling chain from P3H1 to cell division and immune control
Diving deeper, the researchers uncovered a signaling chain inside tumor cells. When P3H1 was reduced, levels of another protein, Polo-like kinase 1 (PLK1), fell at both the protein and RNA level, in cells, mouse tumors, and patient samples. PLK1 is a key regulator of cell division and is already known to be overactive in many cancers. Using phosphoproteomics, the team showed that PLK1 activity normally boosts a central cancer regulator called β-catenin by modifying it at a specific site needed for entry into the cell nucleus. In P3H1-deficient cells, this activating mark on β-catenin dropped, its nuclear presence waned, and expression of β-catenin target genes fell. Those targets include not only cell-cycle drivers like c-Myc and Cyclin D1, which push cells to divide, but also the very cytokines that draw in tumor-supporting macrophages. Restoring either P3H1 or PLK1 in P3H1-deficient cells revived β-catenin activity, cancer cell proliferation, and macrophage recruitment, underscoring that the P3H1–PLK1–β-catenin axis is a central control hub.
Making chemotherapy hit harder
Because β-catenin signaling and tumor-supporting macrophages have both been implicated in resistance to gemcitabine and other standard regimens, the authors asked whether blocking this axis could improve treatment. They combined gemcitabine (and other standard cocktails) with a PLK1 inhibitor, BI2536, in mice bearing human pancreatic tumors. At a carefully chosen dose that avoided organ toxicity, the combination produced much smaller tumors, fewer dividing cancer cells, and fewer tumor-associated macrophages than chemotherapy alone. Patient-derived pancreatic organoids—mini-tumors grown from surgical samples—showed a similar pattern: knocking down P3H1 slowed their growth, lowered PLK1, and made them more vulnerable to gemcitabine plus nab-paclitaxel, while the PLK1 inhibitor further boosted drug sensitivity.
What this means for future treatment
In simple terms, this work identifies a trio of molecules—P3H1, PLK1, and β-catenin—that together act like an accelerator pedal for pancreatic tumors, driving both runaway cell division and the recruitment of immune cells that help the cancer rather than fight it. Disabling this axis slows tumor growth and makes existing chemotherapy more effective in mice and patient-derived models. While drugs that directly block P3H1 do not yet exist, PLK1 inhibitors are already in development, raising the prospect that, in the future, combining them with standard chemotherapy could offer pancreatic cancer patients more durable control of their disease.
Citation: Bai, P., Liu, C., Fu, C. et al. Targeting Prolyl 3-hydroxylase 1 inhibits pancreatic cancer progression and macrophage immunity. Nat Commun 17, 3913 (2026). https://doi.org/10.1038/s41467-026-70452-w
Keywords: pancreatic cancer, tumor microenvironment, macrophages, β-catenin signaling, targeted chemotherapy