Clear Sky Science · ru
Искусственный (in-silico) поиск генетических вариантов, связанных с хроническим лимфоцитарным лейкозом, для диагностических и терапевтических приложений
Почему важно отслеживать рак в наших генах
Хронический лимфоцитарный лейкоз (ХЛЛ) — самый распространённый лейкоз у взрослых, но для многих пациентов он остаётся неизлечимым. В семьях людей с ХЛЛ риск заболевания заметно выше, что указывает на то, что наследственные изменения в ДНК способствуют возникновению болезни. В этом исследовании поставлен практический вопрос: можно ли с помощью крупномасштабных генетических данных и компьютерного анализа выявить ключевые уязвимые места в нашем геноме, которые запускают ХЛЛ — и затем сопоставить их с уже существующими или перспективными препаратами? Ответы могли бы улучшить раннюю диагностику и указать путь к более точному, менее методом проб и ошибок лечению.
Анализ тысяч геномов
Исследователи начали с объединения суммарных результатов трёх крупных исследований ассоциаций по всему геному, которые сканируют ДНК большого числа людей с ХЛЛ и без него в поисках мелких различий, связанных с риском болезни. Тщательно очистив и объединив эти наборы данных, все на одной эталонной версии генома человека, они увеличили статистическую силу для выявления тонких генетических сигналов. С помощью метаанализа, взвешивающего исследования по точности оценок, они идентифицировали 2357 позиций в ДНК, называемых однонуклеотидными полиморфизмами (SNP), которые были тесно связаны с риском ХЛЛ. Эти сигналы сгруппировались в 40 участков генома, включая 11 регионов, ранее не связавшихся с заболеванием.
От маркеров ДНК к ключевым управляющим генам
Большинство маркеров риска располагались между генами или в некодирующих частях генов, где их эффекты неочевидны. Чтобы интерпретировать их, команда использовала биоинформационные инструменты, связывающие каждый маркер с ближайшими генами или с генами, активность которых в клетках крови меняется при наличии данного маркера. Это сопоставление выделило 60 генов, находящихся в или рядом с зонами риска. Учёные затем спросили, какие из этих генов занимают центральные позиции в сети белок–белковых взаимодействий внутри клетки. Сетевой анализ выделил семь генов — TERT, BCL2, BCL2L11, CFLAR, CASP8, LEF1 и FAS — как узлы, соединяющие множество путей. Независимые базы данных о заболеваниях подтвердили сильные связи этих генов с ХЛЛ, а также с другими кровяными онкологическими и иммунными нарушениями.
Как нарушение программируемой гибели клеток подпитывает лейкоз
При изучении функций этих семи генов проступила ясная тема: большинство из них контролируют судьбу клетки — жить или умирать. Несколько, включая BCL2 и BCL2L11, принадлежат к семейству, балансирующему сигналы выживания и сигналы самоуничтожения в «энергетических станциях» клетки — митохондриях. Другие — FAS, CFLAR и CASP8 — управляют параллельным маршрутом «рецепторов смерти» на поверхности клетки. При ХЛЛ этот механизм смещён в сторону выживания: протеины, поддерживающие выживание, гиперактивны, в то время как проапоптотические партнёры блокированы или снижены. Оставшийся ключевой ген, LEF1, участвует в пути роста, который помогает лейкемическим клеткам продолжать делиться. При проведении анализа обогащения ведущими биологическими темами стали гибель клеток, онкологические пути и иммунная сигнализация, что подкрепляет идею о том, что наследственные варианты склоняют эти регуляторные системы к образованию долгоживущих аномальных B-клеток.
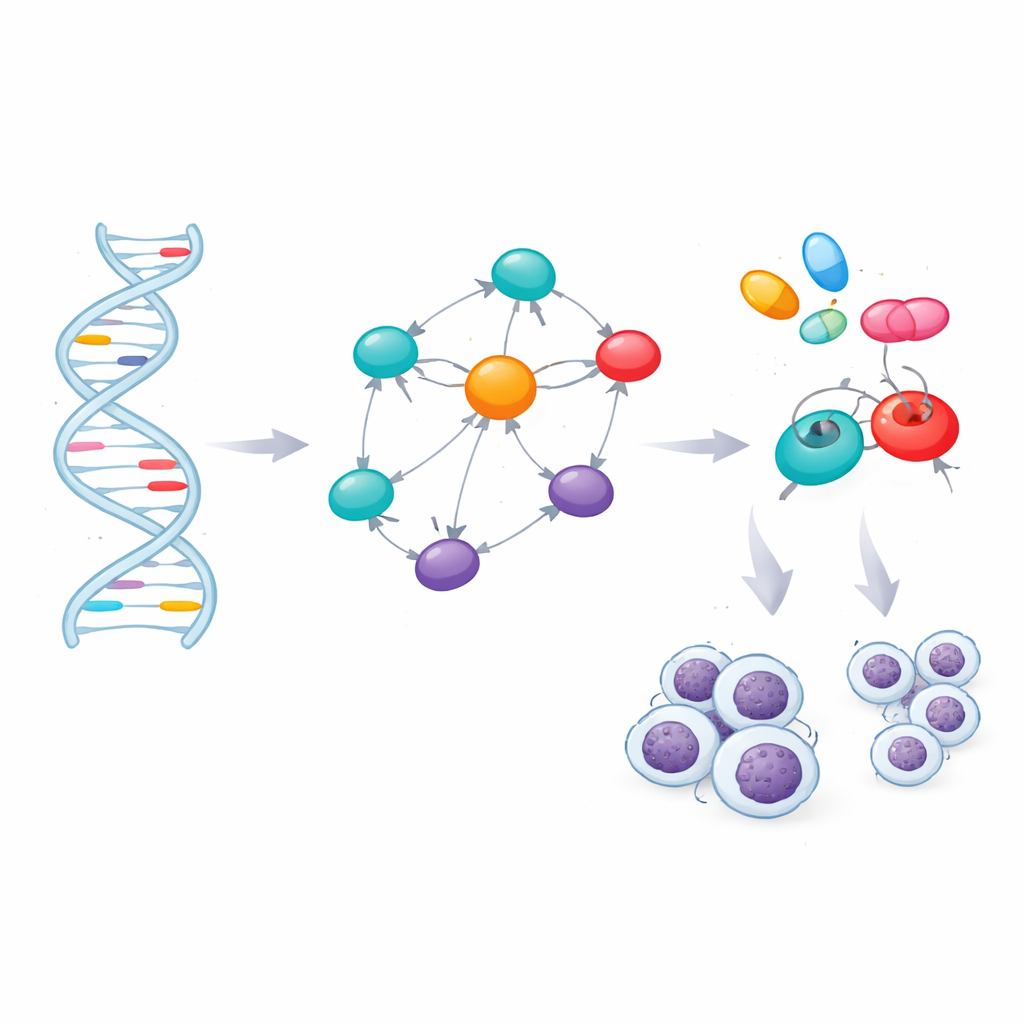
Сопоставление генов с лекарствами
Идентификация ключевых генов открывает путь к таргетной терапии, но только если на эти гены можно воздействовать препаратами. Команда оценила трёхмерные структуры семи белков и обнаружила перспективные карманы, в которые могли бы связываться малые молекулы. Затем они проанализировали библиотеку из 166 существующих или экспериментальных соединений, связанных с ХЛЛ или близкими состояниями, и использовали молекулярное докирование, чтобы увидеть, какие препараты наиболее плотно подходят к ключевым белкам или их основным регуляторам. После ранжирования этих виртуальных взаимодействий кандидаты прошли фильтры стандартных правил «drug-likeness» и компьютерных моделей, прогнозирующих абсорбцию, распределение, метаболизм, выведение и потенциальную токсичность. Такое консервативное отсевание оставило четыре сильных претендента: уже одобренные противораковые препараты ибрутиніб (ibrutinib), селинексор (selinexor), иматиниб (imatinib) и авапритиниб (avapritinib).
Что это может значить для пациентов
Для людей, живущих с ХЛЛ, эта работа пока не меняет повседневную практику, но помогает связать воедино наследственный риск, внутреннюю проводку лейкемических клеток и лекарства, которые могут наиболее эффективно нарушить эту проводку. Два из выделенных препаратов, ибрутиніб и селинексор, уже применяются при ХЛЛ и действуют способами, соответствующими выявленной генетической картине, особенно смещая баланс в сторону гибели раковых клеток. Исследование также предполагает, что иматиниб и авапритиниб, в настоящее время используемые при других видах рака, заслуживают более детального тестирования при ХЛЛ. В более широком смысле подход демонстрирует, как глубокий анализ существующих генетических данных с помощью мощных вычислительных инструментов может выявлять ключевые гены болезни и сужать круг потенциальных терапий, направляя будущие лабораторные эксперименты и клинические испытания к наиболее перспективным мишеням.
Цитирование: Islam, M.S., Mollah, M.H., Ahsan, M.A. et al. In-silico identification of genetic variants associated with chronic lymphocytic leukemia for diagnostic and therapeutic applications. Sci Rep 16, 14545 (2026). https://doi.org/10.1038/s41598-026-45456-7
Ключевые слова: хронический лимфоцитарный лейкоз, генетические варианты риска, пути апоптоза, репозиционирование лекарств, таргетная терапия рака