Clear Sky Science · de
In-silico-Identifizierung genetischer Varianten, die mit chronischer lymphatischer Leukämie in Verbindung stehen, für diagnostische und therapeutische Anwendungen
Warum es wichtig ist, Krebs in unseren Genen nachzuverfolgen
Die chronische lymphatische Leukämie (CLL) ist die häufigste Leukämie im Erwachsenenalter und bleibt dennoch für viele Patienten unheilbar. Familienangehörige von CLL-Patienten haben ein deutlich erhöhtes Risiko, was darauf hindeutet, dass vererbte DNA-Veränderungen die Grundlage für die Erkrankung legen. Diese Studie stellt eine praktische Frage: Lassen sich groß angelegte genetische Daten und Computeranalysen nutzen, um die entscheidenden Schwachstellen in unserer DNA zu identifizieren, die CLL antreiben — und diese Schwachstellen dann mit bestehenden oder vielversprechenden Medikamenten zu verknüpfen? Die Antworten könnten die Früherkennung verbessern und auf präzisere, weniger experimentelle Behandlungsansätze hinweisen.
Blick über Tausende von Genomen
Die Forscher begannen damit, zusammenfassende Ergebnisse aus drei großen genomweiten Assoziationsstudien zu sammeln, die die DNA vieler Personen mit und ohne CLL durchsuchen, um winzige Unterschiede zu finden, die mit dem Krankheitsrisiko verbunden sind. Durch sorgfältiges Bereinigen und Zusammenführen dieser Datensätze, alle auf demselben menschlichen Referenzgenom basierend, erhöhten sie die statistische Power, um subtile genetische Signale zu erkennen. Mithilfe einer Meta-Analyse-Methode, die Studien nach der Präzision ihrer Schätzungen gewichtet, identifizierten sie 2.357 DNA-Positionen, sogenannte Single Nucleotide Polymorphisms, die stark mit CLL assoziiert waren. Diese Signale gruppierten sich in 40 Abschnitte des Genoms, darunter 11 Regionen, die zuvor nicht mit der Krankheit in Verbindung gebracht worden waren.
Von DNA-Markern zu wichtigen Steuerungsgenen
Die meisten Risikomarker lagen zwischen Genen oder in nichtkodierenden Genbereichen, wo ihre Wirkung nicht sofort ersichtlich ist. Um sie zu interpretieren, nutzte das Team bioinformatische Werkzeuge, die jeden Marker mit nahegelegenen Genen oder mit Genen verknüpfen, deren Aktivität in Blutzellen sich ändert, wenn der Marker vorhanden ist. Diese Zuordnung ergab 60 Gene, die sich in oder nahe den Risiko-Regionen befinden. Die Wissenschaftler fragten dann, welche dieser Gene zentrale Positionen im Netzwerk der Protein–Protein-Wechselwirkungen innerhalb der Zellen einnehmen. Eine Netzwerk-Analyse hob sieben Gene hervor — TERT, BCL2, BCL2L11, CFLAR, CASP8, LEF1 und FAS — als Knotenpunkte, die viele Signalwege verbinden. Unabhängige Krankheitsdatenbanken bestätigten starke Verknüpfungen zwischen diesen Genen und CLL sowie anderen Blutkrebserkrankungen und Immunstörungen.
Wie gestörter Zelltod Leukämie antreibt
Bei der Untersuchung der Funktion dieser sieben Gene trat ein klares Muster zutage: Die meisten steuern, ob Zellen überleben oder sterben. Mehrere, darunter BCL2 und BCL2L11, gehören zu einer Familie, die Überlebenssignale mit Selbstzerstörungs-Signalen in den Mitochondrien, den Kraftwerken der Zelle, ausbalanciert. Andere — FAS, CFLAR und CASP8 — regulieren einen parallelen "Death-Rezeptor"-Weg an der Zelloberfläche. Bei CLL ist dieses System in Richtung Überleben verschoben: pro-überlebensfördernde Proteine sind überaktiv, während pro-apoptotische Partner blockiert oder reduziert sind. Das verbleibende Schlüsselgen, LEF1, sitzt in einem Wachstumsweg, der den Leukämiezellen hilft, sich weiter zu teilen. Als die Forscher Anreicherungsanalysen durchführten, waren die wichtigsten biologischen Themen Zelltod, Krebswege und Immun-Signalgebung, was die Idee stützt, dass vererbte Varianten diese Steuerungssysteme zugunsten langlebiger, abnormaler B-Zellen verschieben.
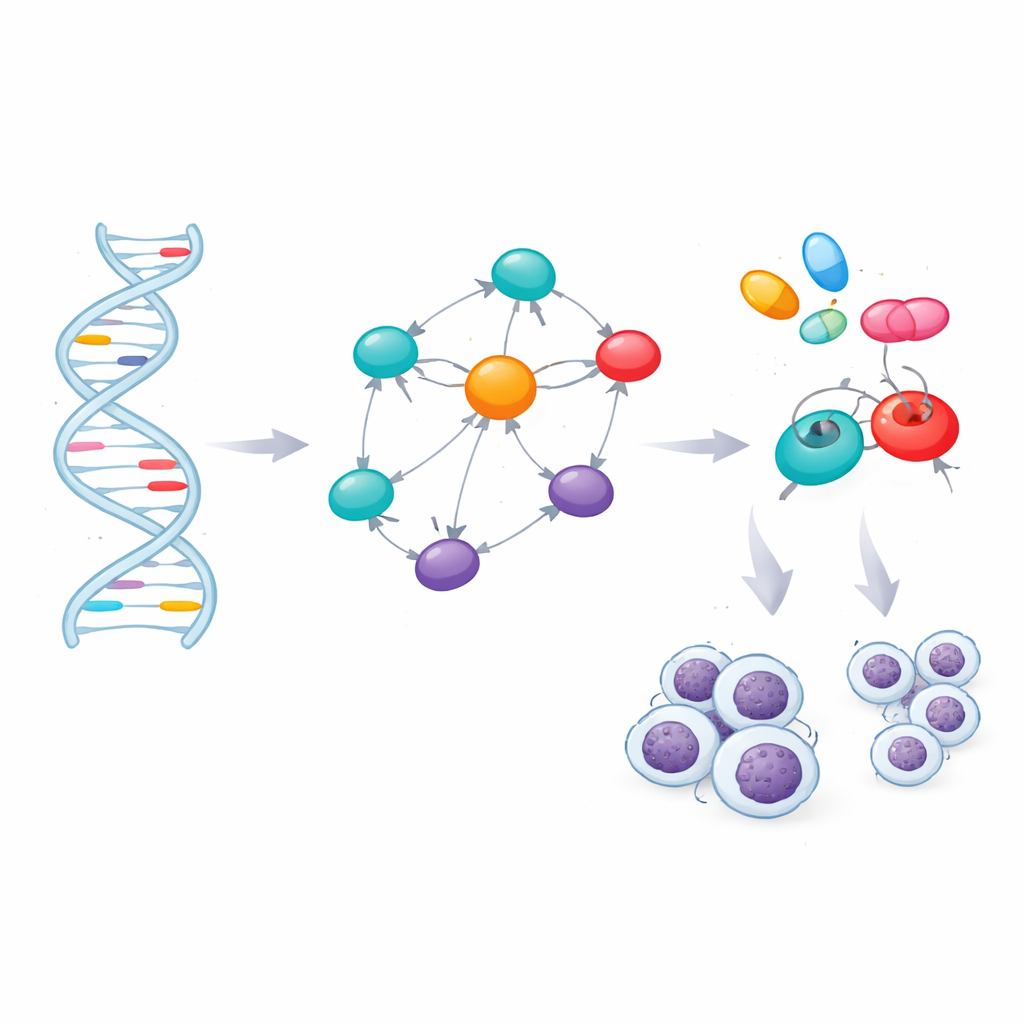
Gene mit Medikamenten abgleichen
Die Identifizierung entscheidender Gene öffnet die Tür zur zielgerichteten Therapie, aber nur, wenn diese Gene durch Medikamente beeinflussbar sind. Das Team bewertete die dreidimensionalen Strukturen der sieben Proteine und fand vielversprechende Taschen, in die kleine Moleküle binden könnten. Anschließend durchsuchten sie eine Bibliothek mit 166 bestehenden oder experimentellen Verbindungen, die mit CLL oder verwandten Erkrankungen in Verbindung stehen, und nutzten molekulares Docking, um zu prüfen, welche Medikamente am besten in die Schlüsselproteine oder deren Hauptregulatoren passen. Nach dem Ranking dieser virtuellen Interaktionen filterten sie die Spitzenkandidaten anhand üblicher Regeln zur "Drug-Likeness" und durch Computermodelle, die vorhersagen, wie gut eine Verbindung absorbiert, verteilt, metabolisiert, ausgeschieden wird und wie toxisch sie sein könnte. Dieses konservative Screening ergab vier starke Anwärter: die bereits zugelassenen Krebsmedikamente Ibrutinib, Selinexor, Imatinib und Avapritinib.
Was das für Patientinnen und Patienten bedeuten könnte
Für Menschen mit CLL ändert diese Arbeit noch nicht die unmittelbare Versorgung, aber sie hilft, die Verbindungen zwischen vererbtem Risiko, der inneren Verdrahtung von Leukämiezellen und den Medikamenten zu verstehen, die diese Verdrahtung am besten stören könnten. Zwei der hervorgehobenen Medikamente, Ibrutinib und Selinexor, werden bereits in CLL eingesetzt und wirken auf Weisen, die zum hier aufgedeckten genetischen Bild passen, insbesondere indem sie das Gleichgewicht wieder zugunsten des Zelltods von Krebszellen verschieben. Die Studie legt außerdem nahe, dass Imatinib und Avapritinib, die derzeit für andere Krebsarten verwendet werden, eine nähere Prüfung in CLL verdienen könnten. Allgemeiner zeigt der Ansatz, wie das Auswerten vorhandener genetischer Daten mit leistungsstarken computergestützten Werkzeugen Schlüsseldiseasegene aufdecken und das Feld potenzieller Therapien eingrenzen kann, um künftige Laborversuche und klinische Studien auf die vielversprechendsten Ziele zu lenken.
Zitation: Islam, M.S., Mollah, M.H., Ahsan, M.A. et al. In-silico identification of genetic variants associated with chronic lymphocytic leukemia for diagnostic and therapeutic applications. Sci Rep 16, 14545 (2026). https://doi.org/10.1038/s41598-026-45456-7
Schlüsselwörter: chronische lymphatische Leukämie, genetische Risikovarianten, Apoptosewege, Arzneimittel-Neuzuordnung, zielgerichtete Krebstherapie