Clear Sky Science · pl
Identyfikacja in silico wariantów genetycznych związanych z przewlekłą białaczką limfocytową do zastosowań diagnostycznych i terapeutycznych
Dlaczego śledzenie nowotworu w naszych genach ma znaczenie
Przewlekła białaczka limfocytowa (CLL) to najczęstsza białaczka u dorosłych, lecz dla wielu pacjentów pozostaje nieuleczalna. Rodziny osób z CLL mają wyraźnie zwiększone ryzyko zachorowania, co sugeruje, że odziedziczone zmiany w DNA przyczyniają się do powstania choroby. W tym badaniu zadano praktyczne pytanie: czy można wykorzystać dane genetyczne na dużą skalę i analizy komputerowe, aby wskazać kluczowe słabe punkty w naszym materiale genetycznym sprzyjające CLL — a następnie powiązać je z istniejącymi lub obiecującymi lekami? Odpowiedzi mogą poprawić wczesną diagnostykę i skierować leczenie ku bardziej precyzyjnym, mniej metodologicznym próbom i błędom.
Analiza tysięcy genomów
Naukowcy zaczęli od zebrania zestawów wyników z trzech dużych badań asocjacyjnych obejmujących cały genom, które przesiewają DNA wielu osób z CLL i bez choroby, aby znaleźć drobne różnice powiązane z ryzykiem. Poprzez staranne oczyszczenie i połączenie tych zestawów danych, wszystkie osadzone względem tego samego ludzkiego genomu referencyjnego, zwiększyli moc statystyczną do wykrywania subtelnych sygnałów genetycznych. Przy użyciu metody metaanalizy ważonej przez precyzję oszacowań zidentyfikowali 2 357 pozycji DNA, zwanych polimorfizmami pojedynczego nukleotydu (SNP), które były silnie powiązane z CLL. Sygnały te zgromadziły się w 40 obszarach genomu, w tym w 11 regionach wcześniej niepowiązanych z tą chorobą.
Od markerów DNA do kluczowych genów kontrolnych
Większość markerów ryzyka znajdowała się między genami lub w niekodujących częściach genów, gdzie ich działanie nie jest oczywiste. Aby je interpretować, zespół wykorzystał narzędzia bioinformatyczne łączące każdy marker z pobliskimi genami lub z genami, których aktywność w komórkach krwi zmienia się w obecności danego markera. To mapowanie dało 60 genów położonych w lub w pobliżu regionów ryzyka. Naukowcy następnie sprawdzili, które z tych genów zajmują centralne pozycje w sieci kontaktów białko–białko wewnątrz komórek. Analiza sieci uwydatniła siedem genów — TERT, BCL2, BCL2L11, CFLAR, CASP8, LEF1 i FAS — jako huby łączące wiele szlaków. Niezależne bazy danych chorób potwierdziły silne powiązania między tymi genami a CLL, a także innymi nowotworami krwi i zaburzeniami układu odpornościowego.
Jak zaburzenia śmierci komórkowej napędzają białaczkę
Gdy zespół przyjrzał się funkcjom tych siedmiu genów, wyłonił się wyraźny motyw: większość z nich kontroluje, czy komórki przeżywają, czy umierają. Kilka, w tym BCL2 i BCL2L11, należy do rodziny białek równoważących sygnały przeżyciowe i sygnały autodestrukcji w mitochondriach, „elektrowniach” komórki. Inne — FAS, CFLAR i CASP8 — regulują równoległą ścieżkę „receptorów śmierci” na powierzchni komórki. W CLL ta aparatura przesuwa się w stronę przeżycia: białka promujące przeżycie są nadaktywne, podczas gdy partnerzy promujący śmierć są zablokowani lub zmniejszeni. Pozostały kluczowy gen, LEF1, uczestniczy w ścieżce wzrostu, która pomaga komórkom białaczkowym w dalszym podziale. Analizy wzbogacenia wykazały, że główne tematy biologiczne to śmierć komórkowa, szlaki nowotworowe i sygnalizacja immunologiczna, co wzmacnia obraz, że odziedziczone warianty przesuwają te systemy kontrolne w stronę długowiecznych, nieprawidłowych limfocytów B.
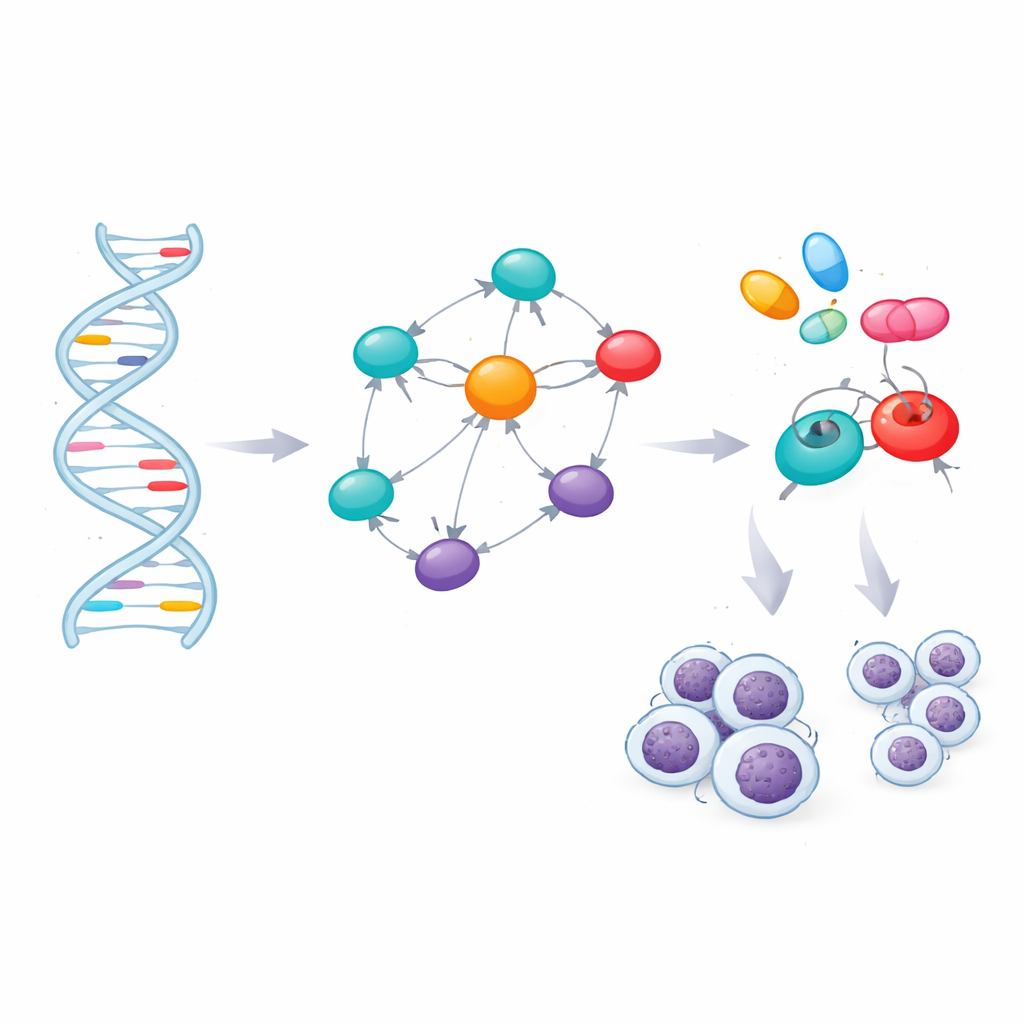
Powiązanie genów z lekami
Identyfikacja kluczowych genów otwiera drzwi do terapii celowanej, ale tylko jeśli te geny można modulować lekami. Zespół ocenił trzywymiarowe struktury siedmiu białek i znalazł obiecujące wnęki, w które mogłyby wnikać małe cząsteczki. Następnie przeszukali bibliotekę 166 istniejących lub eksperymentalnych związków powiązanych z CLL lub pokrewnymi schorzeniami i użyli symulacji dokowania molekularnego, aby sprawdzić, które leki najlepiej dopasowują się do kluczowych białek lub ich głównych regulatorów. Po uszeregowaniu tych wirtualnych interakcji przefiltrowali najlepsze kandydatury przez standardowe reguły „drug-likeness” oraz przez modele komputerowe przewidujące wchłanianie, dystrybucję, metabolizm, wydalanie i potencjalną toksyczność związku. To konserwatywne odfiltrowanie pozostawiło czterech silnych pretendentów: już zatwierdzone leki przeciwnowotworowe ibrutynib, selinexor, imatynib i avaprytynib.
Co to może oznaczać dla pacjentów
Dla osób żyjących z CLL ta praca jeszcze nie zmienia codziennej opieki, ale pomaga połączyć punkty między odziedziczonym ryzykiem, wewnętrznym okablowaniem komórek białaczkowych a lekami, które mogą najskuteczniej to okablowanie zakłócić. Dwa z wyróżnionych leków, ibrutynib i selinexor, są już stosowane w CLL i działają w sposób zgodny z ujawnionym tutaj obrazem genetycznym, szczególnie przez przywracanie równowagi w kierunku śmierci komórek nowotworowych. Badanie sugeruje też, że imatynib i avaprytynib, obecnie stosowane w innych nowotworach, zasługują na bliższe testy w kontekście CLL. Szerzej ujmując, podejście pokazuje, jak wydobywanie istniejących danych genetycznych za pomocą potężnych narzędzi obliczeniowych może ujawnić kluczowe geny choroby i zawęzić pole potencjalnych terapii, kierując przyszłe badania laboratoryjne i próby kliniczne na najbardziej obiecujące cele.
Cytowanie: Islam, M.S., Mollah, M.H., Ahsan, M.A. et al. In-silico identification of genetic variants associated with chronic lymphocytic leukemia for diagnostic and therapeutic applications. Sci Rep 16, 14545 (2026). https://doi.org/10.1038/s41598-026-45456-7
Słowa kluczowe: przewlekła białaczka limfocytowa, genetyczne warianty ryzyka, szlaki apoptozy, ponowne wykorzystanie leków, celowana terapia przeciwnowotworowa