Clear Sky Science · en
In-silico identification of genetic variants associated with chronic lymphocytic leukemia for diagnostic and therapeutic applications
Why tracking cancer in our genes matters
Chronic lymphocytic leukemia (CLL) is the most common adult leukemia, yet it remains incurable for many patients. Families of people with CLL face a sharply higher risk, hinting that inherited DNA changes help set the stage for the disease. This study asks a practical question: can we use large-scale genetic data and computer analysis to pinpoint the key weak spots in our DNA that drive CLL—and then match them to existing or promising drugs? The answers could improve early diagnosis and point to more precise, less trial-and-error treatment.
Looking across thousands of genomes
The researchers started by collecting summary results from three large genome-wide association studies, which scan the DNA of many people with and without CLL to find tiny differences linked to disease risk. By carefully cleaning and merging these datasets, all based on the same human reference genome, they boosted the statistical power to detect subtle genetic signals. Using a meta-analysis method that weighs studies by the precision of their estimates, they identified 2,357 DNA positions, called single nucleotide polymorphisms, that were strongly associated with CLL. These signals clustered into 40 stretches of the genome, including 11 regions that had not previously been tied to the disease.
From DNA markers to key control genes
Most of the risk markers fell between genes or within noncoding parts of genes, where their effects are not obvious. To interpret them, the team used bioinformatics tools that link each marker to nearby genes or to genes whose activity in blood cells changes when that marker is present. This mapping produced 60 genes that sit in or near the risk regions. The scientists then asked which of these genes occupy central positions in the web of protein–protein contacts inside cells. A network analysis highlighted seven genes—TERT, BCL2, BCL2L11, CFLAR, CASP8, LEF1, and FAS—as hubs that connect many pathways. Independent disease databases supported strong links between these genes and CLL, as well as other blood cancers and immune disorders.
How disturbed cell death fuels leukemia
When the team examined what these seven genes do, a clear theme emerged: most of them control whether cells live or die. Several, including BCL2 and BCL2L11, belong to a family that balances survival signals with self-destruct cues in the cell’s powerhouses, the mitochondria. Others—FAS, CFLAR, and CASP8—govern a parallel “death receptor” route at the cell surface. In CLL, this machinery tilts toward survival: pro-survival proteins are overactive, while pro-death partners are blocked or reduced. The remaining key gene, LEF1, sits in a growth pathway that helps leukemia cells keep dividing. When the researchers ran enrichment analyses, the top biological themes were cell death, cancer pathways, and immune signaling, reinforcing the idea that inherited variants nudge these control systems toward long-lived, abnormal B cells.
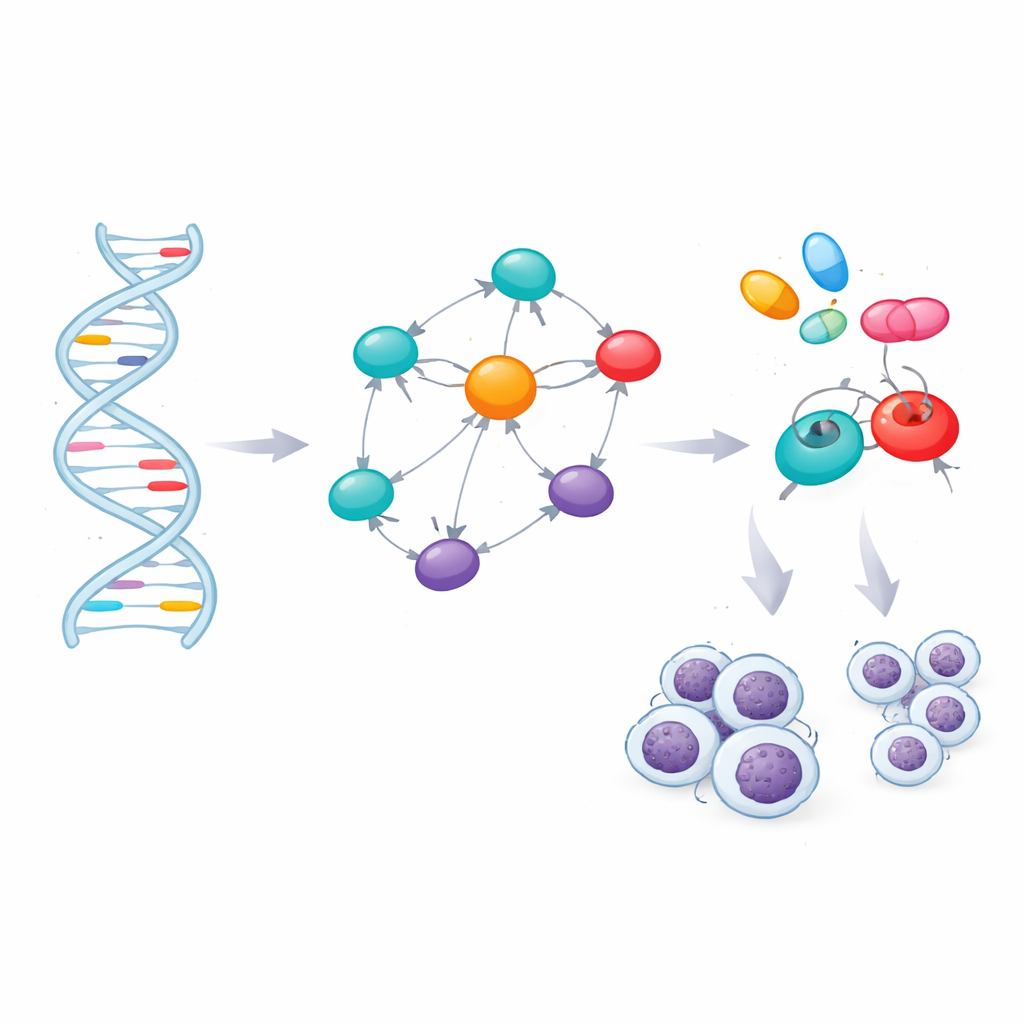
Matching genes to medicines
Identifying crucial genes opens the door to targeted therapy, but only if those genes can be influenced by drugs. The team evaluated the three-dimensional structures of the seven proteins and found promising pockets where small molecules could bind. They then mined a library of 166 existing or experimental compounds connected to CLL or related conditions and used molecular docking simulations to see which drugs fit most snugly into the key proteins or their main regulators. After ranking these virtual interactions, they filtered the top candidates through standard rules for “drug-likeness” and through computer models that predict how well a compound is absorbed, distributed, metabolized, excreted, and how toxic it might be. This conservative screening left four strong contenders: the already approved cancer drugs ibrutinib, selinexor, imatinib, and avapritinib.
What this could mean for patients
For people living with CLL, this work does not yet change day-to-day care, but it helps connect the dots between inherited risk, the internal wiring of leukemia cells, and the medicines that might best disrupt that wiring. Two of the highlighted drugs, ibrutinib and selinexor, are already used in CLL and work in ways that fit the genetic picture uncovered here, especially by tipping the balance back toward cancer cell death. The study also suggests that imatinib and avapritinib, currently used for other cancers, may deserve closer testing in CLL. More broadly, the approach shows how mining existing genetic data with powerful computational tools can reveal key disease genes and narrow the field of potential therapies, guiding future laboratory experiments and clinical trials toward the most promising targets.
Citation: Islam, M.S., Mollah, M.H., Ahsan, M.A. et al. In-silico identification of genetic variants associated with chronic lymphocytic leukemia for diagnostic and therapeutic applications. Sci Rep 16, 14545 (2026). https://doi.org/10.1038/s41598-026-45456-7
Keywords: chronic lymphocytic leukemia, genetic risk variants, apoptosis pathways, drug repurposing, targeted cancer therapy