Clear Sky Science · ru
Патогенные варианты в субъединице загрузчика кохезина MAU2 лежат в основе особого подтипа синдрома Корнелии де Ланж
Когда крошечное изменение гена формирует рост и развитие
Синдром Корнелии де Ланж — редкое состояние, которое влияет на рост, черты лица и способность к обучению. Для многих семей причина остается неизвестной даже после генетического тестирования. В этом исследовании внимание уделено менее изученному гену MAU2: показано, что изменения в этом гене могут вызывать особую форму синдрома Корнелии де Ланж, часто сопровождающуюся низким ростом и малыми размерами головы, но в целом более мягкими нарушениями обучения по сравнению с классическими случаями.
Редкое состояние с множеством проявлений
Синдром Корнелии де Ланж наиболее известен характерной чертой лица, ограничением роста и проблемами развития, но у разных людей он может проявляться по‑разному. У большинства диагностированных обнаруживают изменения в ключевом гене NIPBL, который помогает организовывать упаковку ДНК и её считывание в клетках. Однако значительная часть пациентов не имеет изменений ни в одном из обычных генов, и семьи остаются без ясных ответов. Поскольку MAU2 обычно тесно взаимодействует с NIPBL в одном и том же клеточном механизме, исследователи заподозрили, что тонкие дефекты в MAU2 могут объяснить часть этих нераспознанных случаев.
Связь вариантов MAU2 с клиническими симптомами у людей
Исследователи собрали группу из 18 человек, у всех которых были редкие изменения в одной копии гена MAU2. Эти изменения варьировались от небольших замен аминокислот до преждевременных стоп‑кодонов, приводящих к укорочению белка. Клинике большинство этих людей объединяли два заметных признака: низкий рост и микроцефалия — уменьшенный размер головы по сравнению с ожидаемым для возраста. Многие имели легкие‑умеренные трудности в обучении и некоторые черты лица, перекрывающиеся с синдромом Корнелии де Ланж, хотя в целом менее выраженные, чем при классических вариантах, связанных с NIPBL. По установленной клинической шкале для этого синдрома трое соответствовали критериям «классического» синдрома Корнелии де Ланж, несколько — попали в «неклассическую» категорию, а у некоторых проявления были настолько мягкими, что без генетического тестирования их, вероятно, не распознали бы.
Как дефектный MAU2 нарушает организацию генома
Внутри клеток MAU2 тесно взаимодействует с NIPBL, чтобы «загружать» на ДНК кольцевой белковый комплекс кохезин. Этот комплекс помогает складывать геном и тонко настраивать, какие гены включены или выключены. Команда проверила, как разные варианты MAU2 влияют на это взаимодействие. Многие вариантов, которые удаляли или меняли небольшой участок белка, ослабляли его связь с NIPBL, не разрушая при этом ни один из белков полностью. Другие варианты, вводившие сдвиги рамки считывания или ранние стоп‑кодоны, сокращали уровень MAU2 примерно наполовину — состояние, известное как гаплонедостаточность. В клетках из одной семьи с таким укорочивающим вариантом падение уровня MAU2 также снижало уровень NIPBL, что подчёркивает, насколько плотно эти два партнёра зависят друг от друга для стабильности.
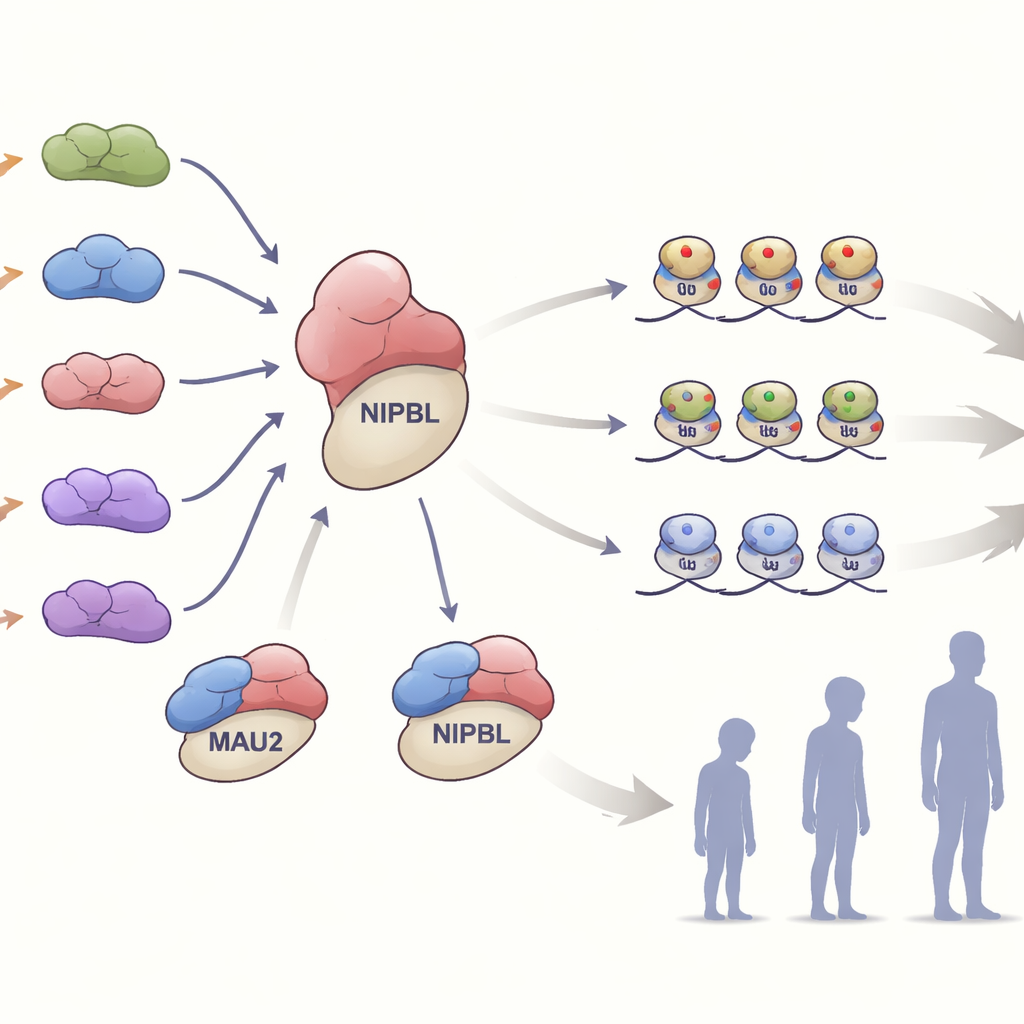
Чтение эпигенетических отпечатков
Исследование вышло за пределы последовательности ДНК и изучило «эпигенетические» метки, которые располагаются поверх генома. В образцах крови поражённых людей учёные измеряли метилирование ДНК — химический маркер, часто отражающий регуляцию генов. У большинства носителей вариантов MAU2 наблюдался профиль метилирования, тесно совпадающий с известной сигнатурой синдрома Корнелии де Ланж, что обеспечивает сильное молекулярное доказательство того, что нарушение MAU2 сходится на том же патологическом пути, что и NIPBL. Интригующим было открытие двух специфичных для MAU2 сигнатур метилирования, которые не полностью перекрывались с классическим рисунком. Некоторые люди, у которых отсутствовала обычная сигнатура Корнелии де Ланж, всё же имели одну из этих MAU2‑специфичных профилей и проявляли более мягкие симптомы, что указывает на то, что частичное нарушение MAU2 может оставлять собственный распознаваемый эпигенетический след.
Модель на мышах отражает человеческие изменения роста и мозга
Чтобы понять, как потеря MAU2 влияет на целый организм, исследователи изучали мышей, генетически модифицированных так, чтобы иметь только одну рабочую копию гена Mau2, отражая человеческую ситуацию. Мыши, полностью лишённые Mau2, не выживали, что подчёркивает жизненно важную роль гена. Выжившие «половинные» мыши были немного ниже по росту, чем их нормальные сиблинги, и имели меньшие по объёму мозги с сокращением отдельных регионов, а также увеличенные заполненные жидкостью полости. Эти находки перекликаются с низким ростом, микроцефалией и тонкими аномалиями мозга у людей с вариантами MAU2, усиливая связь между геном, развитием мозга и внешними признаками.
Что это значит для семей и диагностики
В совокупности данные человека и мыши чётко устанавливают MAU2 как ген, способный вызывать синдром Корнелии де Ланж и близкие состояния. Изменения, которые сильно нарушают партнёрство MAU2–NIPBL, как правило, приводят к появлению полной эпигенетической сигнатуры Корнелии де Ланж и более узнаваемых черт лица, в то время как варианты с более мягким эффектом могут вызывать менее выраженный низкий рост и малый размер головы с только MAU2‑специфическими молекулярными отпечатками. Для семей это означает, что включение MAU2 в диагностические панели и рассмотрение эпигенетического профилирования могут помочь объяснить ранее нераспознанные случаи, а также подчёркивает, как даже небольшие нарушения в механизмах организации генома способны влиять на рост и развитие.
Цитирование: Parenti, I., Hesters, A., Gil-Salvador, M. et al. Pathogenic variants in the cohesin loader subunit MAU2 underlie a distinct Cornelia de Lange Syndrome subtype. Nat Commun 17, 3036 (2026). https://doi.org/10.1038/s41467-026-71177-6
Ключевые слова: синдром Корнелии де Ланж, ген MAU2, загрузчик кохезина, метилирование ДНК, расстройства нейроразвития