Clear Sky Science · es
Variantes patogénicas en la subunidad cargadora de cohesina MAU2 subyacen a un subtipo distinto del síndrome de Cornelia de Lange
Cuando un pequeño cambio en un gen modela el crecimiento y el desarrollo
El síndrome de Cornelia de Lange es una condición rara que afecta el crecimiento, los rasgos faciales y el aprendizaje. Para muchas familias, incluso después de las pruebas genéticas, la causa sigue siendo un misterio. Este estudio se centra en un gen menos conocido llamado MAU2 y muestra que las alteraciones en este gen pueden provocar una forma distinta del síndrome de Cornelia de Lange, a menudo con talla baja y tamaño de cabeza pequeño, pero con dificultades de aprendizaje generalmente más leves que en los casos clásicos.
Una condición rara con muchas caras
El síndrome de Cornelia de Lange es más conocido por su apariencia facial característica, la restricción del crecimiento y los desafíos del desarrollo, pero puede manifestarse de formas muy diferentes entre unas personas y otras. La mayoría de los individuos diagnosticados presentan alteraciones en un gen clave llamado NIPBL, que ayuda a organizar cómo se empaqueta y se lee el ADN dentro de las células. Sin embargo, una fracción notable de pacientes carece de variaciones en cualquiera de los genes habituales, dejando a las familias sin respuestas claras. Dado que MAU2 normalmente actúa de la mano con NIPBL en la misma maquinaria celular, los investigadores sospecharon que fallos sutiles en MAU2 podrían explicar algunos de estos casos no resueltos.
Vinculando las variantes de MAU2 con los síntomas humanos
Los investigadores reunieron a un grupo de 18 individuos que portaban variantes raras en una copia del gen MAU2. Estas variantes iban desde cambios pequeños en un solo aminoácido de la proteína hasta señales de parada prematuras que la truncan. Clínicamente, la mayoría de estos individuos compartían dos rasgos destacados: talla baja y microcefalia, es decir, un tamaño de cabeza menor al esperado para la edad. Muchos presentaban dificultades de aprendizaje de leves a moderadas y algunos rasgos faciales solapados con el síndrome de Cornelia de Lange, aunque generalmente menos pronunciados que en los casos típicos relacionados con NIPBL. Usando un sistema de puntuación clínica establecido para el síndrome, tres personas cumplían los criterios de Cornelia de Lange “clásico”, varias más encajaban en un rango “no clásico” y unas pocas eran tan leves que probablemente no se habrían reconocido sin la prueba genética.
Cómo el MAU2 defectuoso altera la organización del genoma
Dentro de las células, MAU2 forma una asociación estrecha con NIPBL para cargar un complejo proteico en forma de anillo llamado cohesina sobre el ADN. Este complejo ayuda a plegar el genoma y afinar qué genes se activan o se silencian. El equipo evaluó cómo las distintas variantes de MAU2 afectaban esta asociación. Muchas de las variantes que simplemente eliminaban o alteraban un pequeño tramo de la proteína debilitaban su unión a NIPBL sin destruir ninguna de las dos proteínas por completo. Otras variantes, que introducían cambios en el marco de lectura o señales de parada tempranas, reducían los niveles de MAU2 aproximadamente a la mitad, una situación conocida como haploinsuficiencia. En células de una familia con una variante truncante, la reducción de la proteína MAU2 también disminuyó los niveles de NIPBL, lo que subraya la dependencia mutua de ambos socios para su estabilidad.
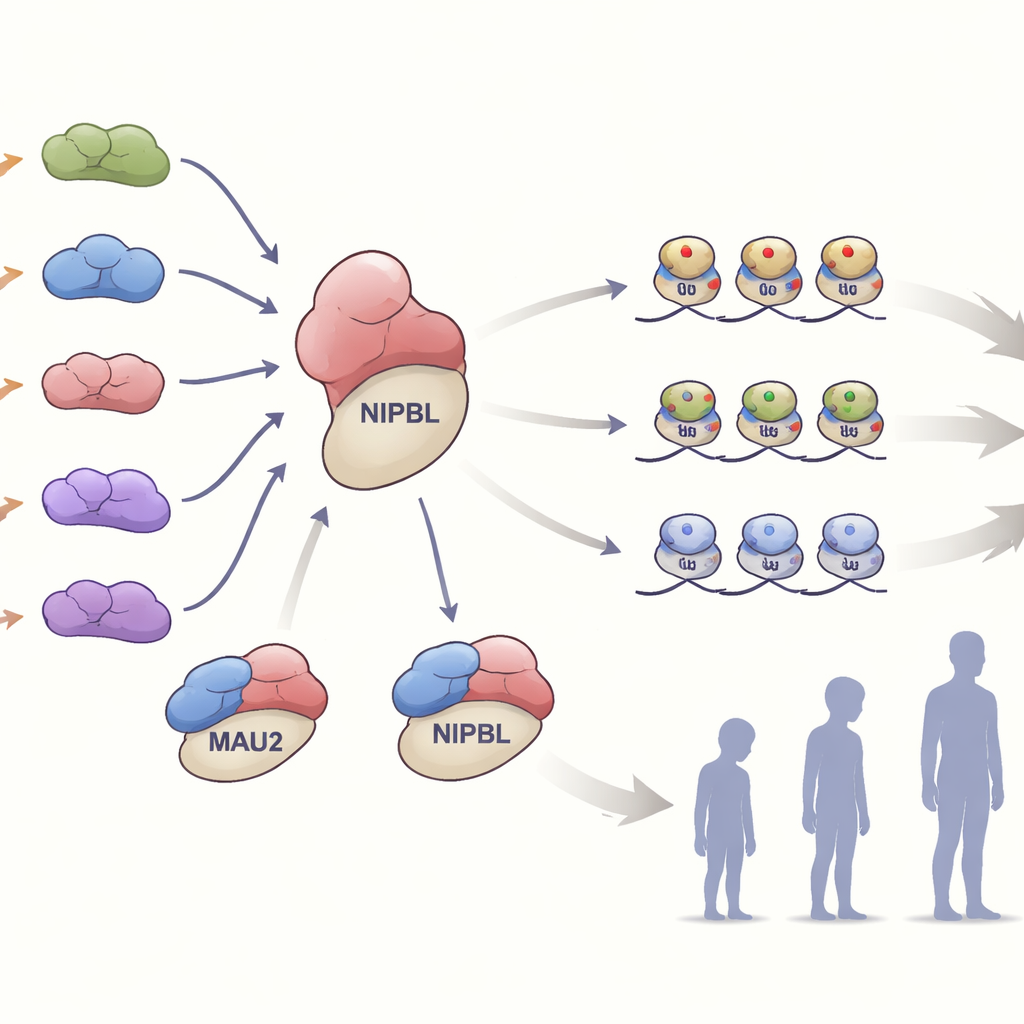
Leyendo las huellas epigenéticas
El estudio fue más allá de la secuencia de ADN para examinar las marcas “epigenéticas” que se superponen al genoma. En células sanguíneas de individuos afectados, los investigadores midieron la metilación del ADN, una etiqueta química que a menudo refleja cómo se regulan los genes. La mayoría de los portadores de variantes en MAU2 mostraron un patrón de metilación que coincidía estrechamente con la firma conocida del síndrome de Cornelia de Lange, aportando una fuerte evidencia molecular de que la disrupción de MAU2 converge en la misma vía patológica que NIPBL. De forma intrigante, el equipo también descubrió dos firmas de metilación específicas de MAU2 que no se solapaban completamente con el patrón clásico. Algunos individuos que carecían de la firma habitual de Cornelia de Lange aún presentaban uno de estos perfiles específicos de MAU2 y tenían síntomas más leves, lo que sugiere que la alteración parcial de MAU2 puede dejar su propia huella epigenética reconocible.
Un modelo murino refleja los cambios en crecimiento y cerebro humanos
Para ver cómo la pérdida de MAU2 afecta a un organismo completo, los investigadores estudiaron ratones diseñados para portar solo una copia funcional del gen Mau2, reflejando la situación humana. Los ratones que carecían totalmente de Mau2 no sobrevivieron, lo que destaca la esencialidad del gen. Los ratones con dosis reducida fueron algo más bajos que sus camadas normales y tenían cerebros más pequeños con regiones específicas reducidas en tamaño, junto con espacios llenos de líquido agrandados. Estos hallazgos recuerdan la talla baja, la microcefalia y las sutiles anomalías cerebrales observadas en personas con variantes en MAU2, reforzando el vínculo entre el gen, el desarrollo cerebral y los rasgos externos.
Qué supone esto para las familias y el diagnóstico
En conjunto, los datos humanos y murinos establecen firmemente a MAU2 como un gen capaz de causar el síndrome de Cornelia de Lange y condiciones estrechamente relacionadas. Las alteraciones que perturban de forma intensa la asociación MAU2–NIPBL tienden a producir la firma epigenética completa de Cornelia de Lange y rasgos faciales más reconocibles, mientras que las variantes con efectos más suaves pueden conducir a una talla baja y microcefalia más leves con solo huellas moleculares específicas de MAU2. Para las familias, esto significa que añadir MAU2 a los paneles diagnósticos y considerar el perfilado epigenético puede ayudar a explicar casos previamente no resueltos, y subraya cómo incluso pequeñas perturbaciones en la maquinaria que organiza el genoma pueden propagarse para moldear el crecimiento y el desarrollo.
Cita: Parenti, I., Hesters, A., Gil-Salvador, M. et al. Pathogenic variants in the cohesin loader subunit MAU2 underlie a distinct Cornelia de Lange Syndrome subtype. Nat Commun 17, 3036 (2026). https://doi.org/10.1038/s41467-026-71177-6
Palabras clave: Síndrome de Cornelia de Lange, gen MAU2, cargador de cohesina, metilación del ADN, trastornos del neurodesarrollo