Clear Sky Science · fr
Les variantes pathogènes de la sous-unité chargeuse de cohésine MAU2 sous-tendent un sous-type distinct du syndrome de Cornelia de Lange
Quand une minuscule modification génétique façonne la croissance et le développement
Le syndrome de Cornelia de Lange est une affection rare qui affecte la croissance, les traits du visage et les capacités d’apprentissage. Pour de nombreuses familles, même après des tests génétiques, la cause reste inconnue. Cette étude se concentre sur un gène moins connu nommé MAU2 et montre que des altérations de ce gène peuvent provoquer une forme distincte du syndrome de Cornelia de Lange, souvent caractérisée par une petite taille et une microcéphalie mais des troubles d’apprentissage généralement plus légers que dans les cas classiques.
Une affection rare aux nombreux visages
Le syndrome de Cornelia de Lange est surtout connu pour son apparence faciale caractéristique, le retard de croissance et les difficultés du développement, mais son expression peut varier fortement d’une personne à l’autre. La plupart des personnes diagnostiquées présentent des altérations d’un gène clé nommé NIPBL, qui contribue à organiser la façon dont l’ADN est emballé et lu dans les cellules. Pourtant, une part notable des patients ne présente pas de variation dans les gènes habituellement impliqués, laissant les familles sans explication claire. Étant donné que MAU2 travaille normalement en étroite coopération avec NIPBL au sein de la même machinerie cellulaire, les chercheurs ont supposé que des anomalies subtiles de MAU2 pourraient rendre compte de certains de ces cas inexpliqués.
Relier les variantes de MAU2 aux symptômes humains
Les chercheurs ont rassemblé un groupe de 18 personnes portant toutes des variations rares sur une copie du gène MAU2. Ces altérations allaient de petites substitutions d’un acide aminé à des signaux d’arrêt prématurés qui tronquent la protéine. Sur le plan clinique, la plupart partageaient deux caractéristiques marquantes : une petite taille et une microcéphalie, c’est-à-dire un périmètre crânien plus petit que prévu pour l’âge. Beaucoup présentaient des difficultés d’apprentissage légères à modérées et certains traits faciaux qui se recoupaient avec le syndrome de Cornelia de Lange, bien que généralement moins marqués que dans les cas typiques liés à NIPBL. En utilisant un système de score clinique établi pour le syndrome, trois personnes répondaient aux critères d’un syndrome de Cornelia de Lange « classique », plusieurs autres se situaient dans une catégorie « non classique », et quelques-uns étaient si légers qu’ils n’auraient probablement pas été reconnus sans le test génétique.
Comment MAU2 défaillant perturbe l’organisation du génome
À l’intérieur des cellules, MAU2 forme un partenariat étroit avec NIPBL pour charger sur l’ADN un complexe protéique en forme d’anneau appelé cohésine. Ce complexe aide à plier le génome et à affiner quels gènes sont activés ou réprimés. L’équipe a testé comment les différentes variantes de MAU2 affectaient ce partenariat. Beaucoup de variantes qui modifiaient ou supprimaient une petite portion de la protéine affaiblissaient son interaction avec NIPBL sans détruire l’une ou l’autre des protéines. D’autres variantes, introduisant des décalages du cadre de lecture ou des codons stop précoces, réduisaient les niveaux de MAU2 d’environ moitié, une situation connue sous le nom d’haploinsuffisance. Dans des cellules d’une famille porteuse d’une variante tronquante, la baisse de la protéine MAU2 entraînait aussi une diminution des niveaux de NIPBL, soulignant à quel point les deux partenaires dépendent l’un de l’autre pour leur stabilité.
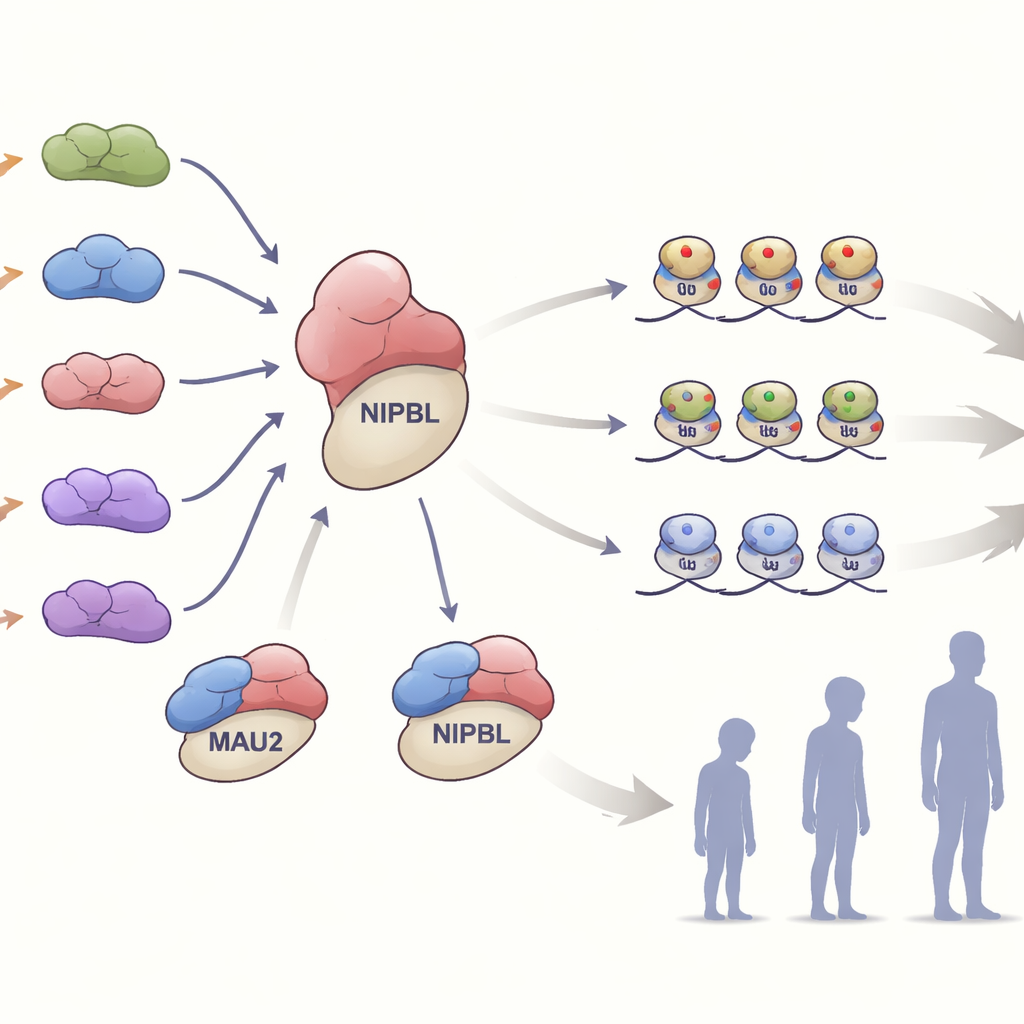
Lire les empreintes épigénétiques
L’étude est allée au-delà de la séquence d’ADN pour examiner les marques « épigénétiques » qui s’ajoutent au génome. Dans les cellules sanguines des individus affectés, les chercheurs ont mesuré la méthylation de l’ADN, un marquage chimique qui reflète souvent la régulation des gènes. La plupart des porteurs de variantes MAU2 présentaient un profil de méthylation proche de la signature connue du syndrome de Cornelia de Lange, fournissant une forte preuve moléculaire que la perturbation de MAU2 converge vers la même voie pathologique que NIPBL. Fait intéressant, l’équipe a aussi identifié deux signatures de méthylation spécifiques à MAU2 qui ne recoupaient pas entièrement le motif classique. Certaines personnes dépourvues de la signature classique présentaient néanmoins l’un de ces profils spécifiques à MAU2 et avaient des symptômes plus légers, ce qui suggère qu’une perturbation partielle de MAU2 peut laisser une empreinte épigénétique reconnaissable propre.
Un modèle murin reflète les changements de croissance et cérébraux humains
Pour évaluer les effets de la perte de MAU2 à l’échelle d’un organisme entier, les chercheurs ont étudié des souris génétiquement modifiées pour ne porter qu’une seule copie fonctionnelle du gène Mau2, reproduisant la situation humaine. Les souris totalement dépourvues de Mau2 ne survivaient pas, ce qui souligne le caractère essentiel du gène. Les animaux hétérozygotes étaient légèrement plus petits que leurs congénères normaux et présentaient des cerveaux plus petits avec certaines régions réduites en volume, ainsi que des espaces remplis de liquide élargis. Ces observations font écho à la petite taille, à la microcéphalie et aux anomalies cérébrales subtiles observées chez les personnes porteuses de variantes MAU2, renforçant le lien entre le gène, le développement cérébral et les caractéristiques externes.
Ce que cela signifie pour les familles et le diagnostic
Pris ensemble, les données humaines et murines établissent solidement MAU2 comme un gène capable de provoquer le syndrome de Cornelia de Lange et des affections étroitement apparentées. Les altérations qui perturbent fortement le partenariat MAU2–NIPBL tendent à produire la signature épigénétique complète du syndrome et des traits faciaux plus reconnaissables, tandis que les variantes aux effets plus doux peuvent entraîner une petite taille et une microcéphalie plus modérées avec uniquement des empreintes moléculaires spécifiques à MAU2. Pour les familles, cela signifie qu’ajouter MAU2 aux panels de diagnostic et envisager un profilage épigénétique peut aider à résoudre des cas auparavant inexpliqués, et cela souligne comment même de petites perturbations de la machinerie d’organisation du génome peuvent se répercuter sur la croissance et le développement.
Citation: Parenti, I., Hesters, A., Gil-Salvador, M. et al. Pathogenic variants in the cohesin loader subunit MAU2 underlie a distinct Cornelia de Lange Syndrome subtype. Nat Commun 17, 3036 (2026). https://doi.org/10.1038/s41467-026-71177-6
Mots-clés: syndrome de Cornelia de Lange, gène MAU2, chargeur de cohésine, méthylation de l’ADN, troubles du neurodéveloppement