Clear Sky Science · pl
Patogenne warianty podjednostki ładującej kohezynę MAU2 leżą u podstaw odrębnego podtypu zespołu Cornelii de Lange
Kiedy drobna zmiana w genie kształtuje wzrost i rozwój
Zespół Cornelii de Lange to rzadka choroba wpływająca na wzrost, rysy twarzy i uczenie się. Dla wielu rodzin, nawet po badaniach genetycznych, przyczyna pozostaje nieznana. W tym badaniu skupiono się na mniej znanym genie o nazwie MAU2 i wykazano, że zmiany w tym genie mogą powodować odrębną postać zespołu Cornelii de Lange, często z niskim wzrostem i małą głową, ale zazwyczaj z łagodniejszymi trudnościami w uczeniu się niż w klasycznych przypadkach.
Rzadka choroba o wielu obliczach
Zespół Cornelii de Lange jest najbardziej znany z charakterystycznego wyglądu twarzy, zahamowania wzrostu i problemów rozwojowych, lecz może wyglądać bardzo różnie u poszczególnych osób. U większości rozpoznanych pacjentów wykrywa się zmiany w kluczowym genie NIPBL, który pomaga organizować, jak DNA jest pakowany i odczytywany w komórkach. Niemniej znaczna część pacjentów nie ma wariantów w typowych genach, pozostawiając rodziny bez jasnej odpowiedzi. Ponieważ MAU2 normalnie współpracuje z NIPBL w tym samym aparacie komórkowym, badacze przypuszczali, że subtelne usterki w MAU2 mogą wyjaśnić niektóre z tych nierozpoznanych przypadków.
Powiązanie wariantów MAU2 z objawami u ludzi
Naukowcy zgromadzili grupę 18 osób, które wszystkie nosiły rzadkie zmiany w jednej kopii genu MAU2. Zmiany te obejmowały od drobnych zamian pojedynczych aminokwasów po sygnały przedwczesnego zakończenia, które skracały białko. Klinicznie większość tych osób miała dwa wyraźne cechy wspólne: niski wzrost i mikrocefalię, czyli rozmiar głowy mniejszy niż oczekiwany dla wieku. Wielu miało łagodne do umiarkowanych trudności w uczeniu się oraz niektóre cechy twarzy nakładające się na zespół Cornelii de Lange, choć zazwyczaj mniej nasilone niż w typowych przypadkach związanych z NIPBL. Przy użyciu ustalonego systemu punktacji klinicznej trzy osoby spełniały kryteria „klasycznego” zespołu Cornelii de Lange, kilka kolejnych mieściło się w zakresie „nieklasycznym”, a kilku było tak łagodnych, że prawdopodobnie nie zostałyby rozpoznane bez badań genetycznych.
Jak wadliwe MAU2 zaburza organizację genomu
W komórkach MAU2 tworzy bliskie partnerstwo z NIPBL, aby ładować pierścieniowy kompleks białkowy zwany kohezyną na DNA. Kompleks ten pomaga składać genom i precyzować, które geny są włączane lub wyłączane. Zespół sprawdził, jak różne zmiany w MAU2 wpływają na to partnerstwo. Wiele wariantów, które jedynie usuwały lub modyfikowały niewielki fragment białka, osłabiało jego wiązanie z NIPBL, nie niszcząc jednak bezpośrednio żadnego z białek. Inne warianty, wprowadzające przesunięcia ramki odczytu lub wczesne sygnały stop, zmniejszały poziomy MAU2 mniej więcej o połowę — stan znany jako haploinsuficencja. W komórkach pochodzących z jednej rodziny z takim wariantem truncującym spadek poziomu białka MAU2 towarzyszył również obniżony poziom NIPBL, podkreślając ścisłe uzależnienie obu partnerów od siebie dla stabilności.
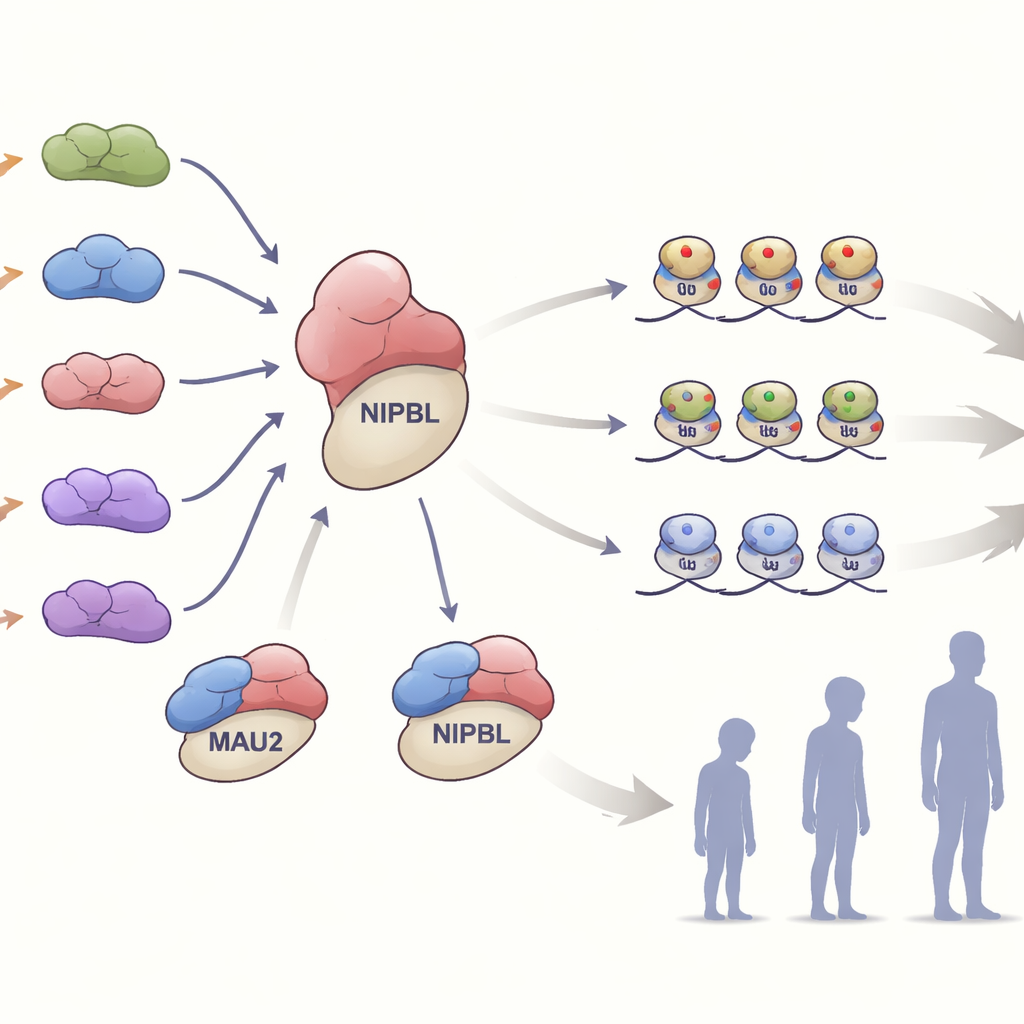
Odczytywanie epigenetycznych odcisków palca
Badanie poszło dalej niż sekwencja DNA i obejrzało „epigenetyczne” znaki nakładane na genom. W komórkach krwi osób dotkniętych badacze zmierzyli metylację DNA, chemiczny znacznik, który często odzwierciedla sposób regulacji genów. Większość nosicieli wariantów MAU2 wykazała wzorzec metylacji bardzo podobny do znanego sygnaturowego profilu zespołu Cornelii de Lange, dostarczając silnych dowodów molekularnych, że zaburzenie MAU2 zbiega się na tę samą ścieżkę chorobową co NIPBL. Co ciekawe, zespół odkrył również dwie specyficzne sygnatury metylacyjne związane z MAU2, które nie pokrywały się całkowicie z klasycznym wzorcem. Niektórzy pacjenci pozbawieni typowej sygnatury Cornelii de Lange mieli nadal jedną z tych specyficznych profili MAU2 i wykazywali łagodniejsze objawy, co sugeruje, że częściowe zaburzenie MAU2 może zostawić własny, rozpoznawalny ślad epigenetyczny.
Model myszy odzwierciedla zmiany wzrostu i mózgu u ludzi
Aby sprawdzić, jak utrata MAU2 wpływa na cały organizm, badacze zbadali myszy zmodyfikowane tak, aby miały tylko jedną czynną kopię genu Mau2, naśladując sytuację ludzką. Myszy całkowicie pozbawione Mau2 nie przeżywały, co pokazuje, jak istotny jest ten gen. Myszy z połowiczną dawką były nieco niższe od normalnych rodzeństwa i miały mniejsze mózgi z obniżonymi rozmiarami konkretnych obszarów oraz powiększonymi przestrzeniami wypełnionymi płynem. Odkrycia te odzwierciedlają niski wzrost, mikrocefalię i subtelne anomalie mózgowe obserwowane u ludzi z wariantami MAU2, wzmacniając związek między genem, rozwojem mózgu a cechami zewnętrznymi.
Co to oznacza dla rodzin i diagnostyki
W sumie dane ludzkie i mysie wyraźnie ustanawiają MAU2 jako gen, który może powodować zespół Cornelii de Lange i pokrewne schorzenia. Zmiany, które silnie zaburzają partnerstwo MAU2–NIPBL, mają tendencję do wywoływania pełnej epigenetycznej sygnatury Cornelii de Lange i bardziej rozpoznawalnych cech twarzy, podczas gdy warianty o łagodniejszym działaniu mogą prowadzić do łagodniejszego niskiego wzrostu i małej głowy z jedynie specyficznymi molekularnymi odciskami MAU2. Dla rodzin oznacza to, że dodanie MAU2 do paneli diagnostycznych oraz rozważenie profilowania epigenetycznego może pomóc wyjaśnić wcześniej nierozwiązane przypadki, i podkreśla, jak nawet niewielkie zaburzenia w maszynerii organizującej genom mogą rozlać się na zewnątrz, kształtując wzrost i rozwój.
Cytowanie: Parenti, I., Hesters, A., Gil-Salvador, M. et al. Pathogenic variants in the cohesin loader subunit MAU2 underlie a distinct Cornelia de Lange Syndrome subtype. Nat Commun 17, 3036 (2026). https://doi.org/10.1038/s41467-026-71177-6
Słowa kluczowe: zespół Cornelii de Lange, gen MAU2, ładujący kohezynę, metylacja DNA, zaburzenia neurorozwojowe