Clear Sky Science · de
Pathogene Varianten in der Kohesin-Lader-Subeinheit MAU2 verursachen einen eigenständigen Subtyp des Cornelia-de-Lange-Syndroms
Wenn eine winzige Genveränderung Wachstum und Entwicklung prägt
Das Cornelia-de-Lange-Syndrom ist eine seltene Erkrankung, die Wachstum, Gesichtszüge und Lernfähigkeit beeinträchtigt. Für viele Familien bleibt die Ursache selbst nach genetischen Tests unklar. Diese Studie konzentriert sich auf ein weniger bekanntes Gen namens MAU2 und zeigt, dass Veränderungen in diesem Gen eine eigenständige Form des Cornelia-de-Lange-Syndroms verursachen können, häufig begleitet von Kleinwuchs und kleinem Kopfumfang, aber im Allgemeinen milderen Lernproblemen als bei klassischen Fällen.
Eine seltene Erkrankung mit vielen Erscheinungsformen
Das Cornelia-de-Lange-Syndrom ist vor allem für sein charakteristisches Gesicht, Wachstumsverzögerung und Entwicklungsprobleme bekannt, kann aber von Person zu Person sehr unterschiedlich aussehen. Die meisten diagnostizierten Personen tragen Veränderungen in einem Schlüsselgen namens NIPBL, das hilft, die Verpackung und Ablesung der DNA in Zellen zu organisieren. Dennoch bleibt bei einem beträchtlichen Teil der Patienten keine Veränderung in den üblichen Genen nachweisbar, sodass Familien ohne klare Erklärung zurückbleiben. Da MAU2 normalerweise eng mit NIPBL in derselben zellulären Maschinerie zusammenarbeitet, vermuteten die Forscher, dass subtile Fehler in MAU2 einige dieser unerklärten Fälle erklären könnten.
Verknüpfung von MAU2-Varianten mit menschlichen Symptomen
Die Forschenden stellten eine Gruppe von 18 Personen zusammen, die alle seltene Veränderungen in einer Kopie des MAU2-Gens trugen. Diese Veränderungen reichten von einzelnen Aminosäureaustauschen bis zu vorzeitigen Stoppsignalen, die das Protein verkürzen. Klinisch wiesen die meisten dieser Personen zwei auffällige Merkmale gemeinsam auf: Kleinwuchs und Mikrozephalie, also einen kleineren Kopfumfang als für das Alter erwartet. Viele hatten leichte bis mäßige Lernschwierigkeiten und einige Gesichtszüge, die mit dem Cornelia-de-Lange-Syndrom überlappten, wenngleich insgesamt weniger ausgeprägt als bei typischen NIPBL-assoziierten Fällen. Nach einem etablierten klinischen Bewertungssystem für das Syndrom erfüllten drei Personen die Kriterien für das „klassische“ Cornelia-de-Lange, mehrere fielen in einen „nicht-klassischen“ Bereich, und einige waren so mild, dass sie wahrscheinlich ohne genetische Tests nicht erkannt worden wären.
Wie fehlerhaftes MAU2 die Organisation des Genoms stört
In den Zellen bildet MAU2 eine enge Partnerschaft mit NIPBL, um einen ringförmigen Proteinkomplex namens Kohesin auf der DNA zu platzieren. Dieser Komplex hilft, das Genom zu falten und fein zu steuern, welche Gene ein- oder ausgeschaltet werden. Das Team untersuchte, wie die verschiedenen MAU2-Veränderungen diese Partnerschaft beeinflussten. Viele Varianten, die lediglich einen kleinen Abschnitt des Proteins entfernten oder veränderten, schwächten dessen Bindung an NIPBL, ohne eines der Proteine vollständig zu zerstören. Andere Varianten, die Frameshifts oder frühe Stoppsignale einführten, reduzierten die MAU2-Menge ungefähr um die Hälfte – eine Situation, die als Haploinsuffizienz bekannt ist. In Zellen aus einer Familie mit einer solchen truncierenden Variante führte der Rückgang des MAU2-Proteins auch zu geringeren NIPBL-Spiegeln, was die enge Abhängigkeit der beiden Partner für ihre Stabilität unterstreicht.
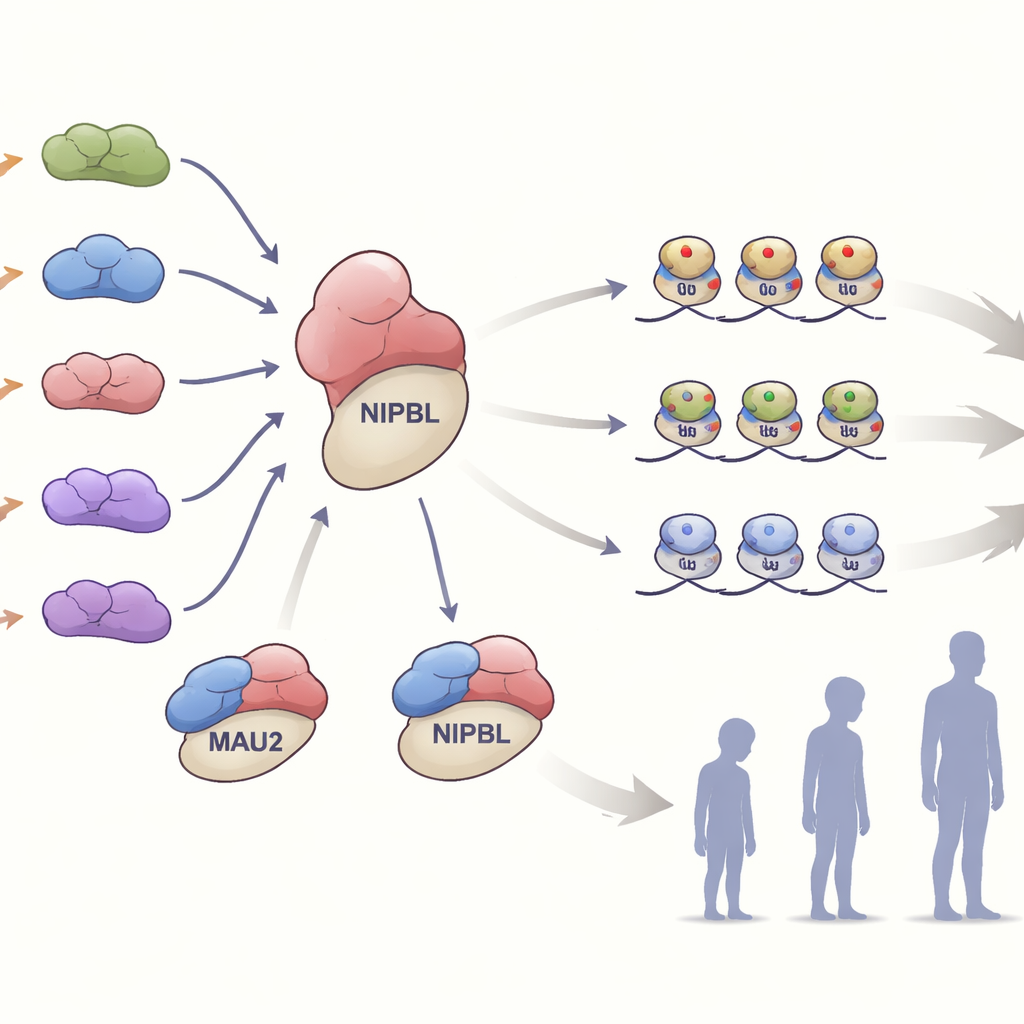
Die epigenetischen Fingerabdrücke lesen
Die Studie ging über die DNA-Sequenz hinaus und untersuchte die „epigenetischen“ Markierungen, die auf dem Genom liegen. In Blutzellen der betroffenen Personen maßen die Forschenden die DNA-Methylierung, einen chemischen Marker, der oft widerspiegelt, wie Gene reguliert werden. Die meisten MAU2-Varianzträger zeigten ein Methylierungsmuster, das eng mit der bekannten Signatur des Cornelia-de-Lange-Syndroms übereinstimmte, und lieferten damit starke molekulare Belege dafür, dass eine Störung von MAU2 auf denselben Krankheitsweg wie NIPBL hinausläuft. Interessanterweise entdeckte das Team zudem zwei MAU2-spezifische Methylierungssignaturen, die nicht vollständig mit dem klassischen Muster überlappten. Einige Personen, die die übliche Cornelia-de-Lange-Signatur nicht zeigten, hatten dennoch eines dieser MAU2-spezifischen Profile und mildere Symptome, was darauf hindeutet, dass eine partielle Störung von MAU2 ihre eigenen erkennbaren epigenetischen Spuren hinterlassen kann.
Ein Mausmodell spiegelt menschliche Wachstums- und Hirnveränderungen wider
Um zu sehen, wie der Verlust von MAU2 einen ganzen Organismus betrifft, untersuchten die Forschenden Mäuse, die so gezüchtet waren, dass sie nur eine funktionierende Kopie des Mau2-Gens trugen – entsprechend der menschlichen Situation. Mäuse, denen Mau2 vollständig fehlte, überlebten nicht, was die essentielle Funktion des Gens unterstreicht. Die überlebenden Halb-Dosis-Mäuse waren etwas kleiner als ihre normalen Wurfgeschwister und hatten kleinere Gehirne mit in bestimmten Regionen reduzierter Größe sowie vergrößerten mit Flüssigkeit gefüllten Räumen. Diese Befunde spiegeln Kleinwuchs, Mikrozephalie und subtile Hirnanomalien bei Menschen mit MAU2-Varianten wider und stärken die Verbindung zwischen Gen, Gehirnentwicklung und beobachtbaren Merkmalen.
Was das für Familien und Diagnostik bedeutet
Zusammen genommen etablieren die menschlichen und Mausdaten MAU2 eindeutig als ein Gen, das das Cornelia-de-Lange-Syndrom und eng verwandte Zustände verursachen kann. Veränderungen, die die MAU2–NIPBL-Partnerschaft stark stören, führen eher zur vollständigen Cornelia-de-Lange-epigenetischen Signatur und zu deutlich erkennbaren Gesichtszügen, während Varianten mit milderen Effekten zu weniger ausgeprägtem Kleinwuchs und kleinem Kopfumfang mit nur MAU2-spezifischen molekularen Fingerabdrücken führen können. Für Familien bedeutet dies, dass die Aufnahme von MAU2 in diagnostische Testpanels und die Erwägung epigenetischer Profilierung dazu beitragen können, zuvor ungeklärte Fälle zu erklären, und es verdeutlicht, wie selbst kleine Störungen in der Organisationsmaschinerie des Genoms weitreichende Auswirkungen auf Wachstum und Entwicklung haben können.
Zitation: Parenti, I., Hesters, A., Gil-Salvador, M. et al. Pathogenic variants in the cohesin loader subunit MAU2 underlie a distinct Cornelia de Lange Syndrome subtype. Nat Commun 17, 3036 (2026). https://doi.org/10.1038/s41467-026-71177-6
Schlüsselwörter: Cornelia-de-Lange-Syndrom, MAU2-Gen, Kohesin-Lader, DNA-Methylierung, neuroentwicklungsstörungen