Clear Sky Science · en
Pathogenic variants in the cohesin loader subunit MAU2 underlie a distinct Cornelia de Lange Syndrome subtype
When a Tiny Gene Change Shapes Growth and Development
Cornelia de Lange syndrome is a rare condition that affects growth, facial features, and learning. For many families, even after genetic testing, the cause remains a mystery. This study focuses on a lesser-known gene called MAU2 and shows that changes in this gene can cause a distinct form of Cornelia de Lange syndrome, often with short stature and small head size but generally milder learning problems than in classic cases.
A Rare Condition with Many Faces
Cornelia de Lange syndrome is best known for its characteristic facial appearance, growth restriction, and developmental challenges, but it can look very different from one person to another. Most diagnosed individuals carry changes in a key gene called NIPBL, which helps organize how DNA is packaged and read inside cells. Yet a sizable fraction of patients lack alterations in any of the usual genes, leaving families without clear answers. Because MAU2 normally works hand in hand with NIPBL in the same cellular machinery, researchers suspected that subtle faults in MAU2 might account for some of these unexplained cases.
Connecting MAU2 Variants to Human Symptoms
The researchers assembled a group of 18 individuals who all carried rare changes in one copy of the MAU2 gene. These changes ranged from small swaps of one building block of the protein to premature stop signals that truncate it. Clinically, most of these individuals shared two prominent traits: short stature and microcephaly, meaning a head size smaller than expected for age. Many had mild to moderate learning difficulties and some facial features overlapping with Cornelia de Lange syndrome, though generally less pronounced than in typical NIPBL-related cases. Using an established clinical scoring system for the syndrome, three people fit the criteria for “classic” Cornelia de Lange, several more fell into a “non-classic” range, and a few were so mild that they likely would not have been recognized without genetic testing.
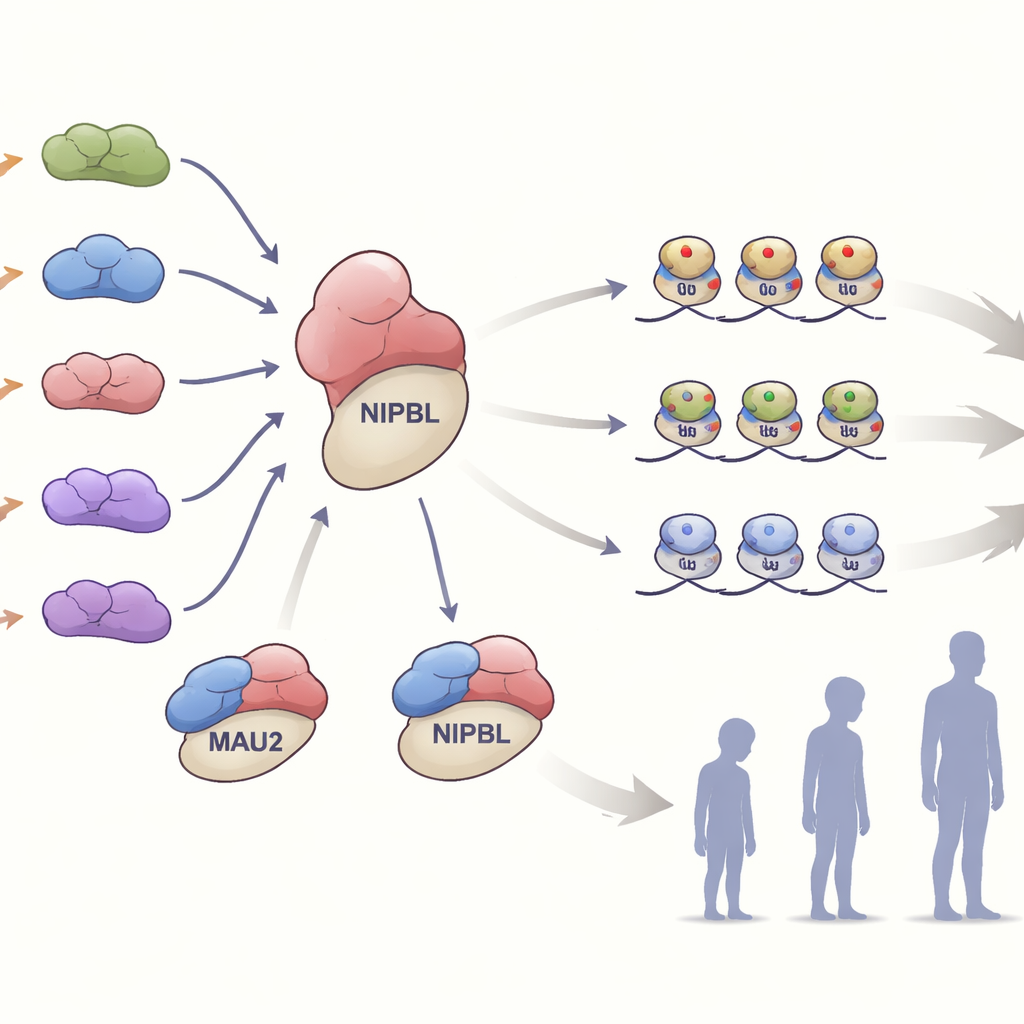
How Faulty MAU2 Disrupts the Genome’s Organization
Inside cells, MAU2 forms a close partnership with NIPBL to load a ring-shaped protein complex called cohesin onto DNA. This complex helps fold the genome and fine-tune which genes are switched on or off. The team tested how the various MAU2 changes affected this partnership. Many of the variants that simply removed or altered a small stretch of the protein weakened its grip on NIPBL without destroying either protein outright. Other variants, which introduced frameshifts or early stop signals, cut MAU2 levels roughly in half, a situation known as haploinsufficiency. In cells from one family with such a truncating variant, the drop in MAU2 protein also reduced NIPBL levels, underscoring how tightly the two partners depend on one another for stability.
Reading the Epigenetic Fingerprints
The study went beyond DNA sequence to examine the “epigenetic” marks that sit on top of the genome. In blood cells from affected individuals, the researchers measured DNA methylation, a chemical tag that often reflects how genes are regulated. Most MAU2 variant carriers showed a methylation pattern closely matching the known signature of Cornelia de Lange syndrome, providing strong molecular evidence that MAU2 disruption converges on the same disease pathway as NIPBL. Intriguingly, the team also discovered two MAU2-specific methylation signatures that did not fully overlap with the classic pattern. Some individuals who lacked the usual Cornelia de Lange signature still carried one of these MAU2-specific profiles and had milder symptoms, suggesting that partial disruption of MAU2 can leave its own recognizable epigenetic trace.
A Mouse Model Mirrors Human Growth and Brain Changes
To see how MAU2 loss affects a whole organism, the researchers studied mice engineered to carry only one working copy of the Mau2 gene, mirroring the human situation. Mice completely lacking Mau2 did not survive, highlighting how essential the gene is. The surviving half-dose mice were slightly shorter than their normal littermates and had smaller brains with specific regions reduced in size, along with enlarged fluid-filled spaces. These findings echo the short stature, microcephaly, and subtle brain anomalies seen in people with MAU2 variants, strengthening the link between gene, brain development, and outward traits.
What This Means for Families and Diagnosis
Taken together, the human and mouse data firmly establish MAU2 as a gene that can cause Cornelia de Lange syndrome and closely related conditions. Changes that strongly disturb the MAU2–NIPBL partnership tend to produce the full Cornelia de Lange epigenetic signature and more recognizable facial features, while variants with gentler effects may lead to milder short stature and small head size with only MAU2-specific molecular fingerprints. For families, this means that adding MAU2 to diagnostic testing panels and considering epigenetic profiling can help explain previously unsolved cases, and it highlights how even small disruptions in the genome’s organizing machinery can ripple outward to shape growth and development.
Citation: Parenti, I., Hesters, A., Gil-Salvador, M. et al. Pathogenic variants in the cohesin loader subunit MAU2 underlie a distinct Cornelia de Lange Syndrome subtype. Nat Commun 17, 3036 (2026). https://doi.org/10.1038/s41467-026-71177-6
Keywords: Cornelia de Lange syndrome, MAU2 gene, cohesin loader, DNA methylation, neurodevelopmental disorders