Clear Sky Science · it
Varianti patogene nella subunità loader della cohesina MAU2 alla base di un sottotipo distinto della sindrome di Cornelia de Lange
Quando una piccola variazione genetica rivela la crescita e lo sviluppo
La sindrome di Cornelia de Lange è una condizione rara che influenza la crescita, i tratti del viso e l’apprendimento. Per molte famiglie, anche dopo i test genetici, la causa resta un mistero. Questo studio si concentra su un gene meno noto chiamato MAU2 e mostra che alterazioni in questo gene possono provocare una forma distinta della sindrome di Cornelia de Lange, spesso caratterizzata da bassa statura e microcefalia ma con difficoltà di apprendimento generalmente più lievi rispetto ai casi classici.
Una condizione rara dai molti volti
La sindrome di Cornelia de Lange è nota soprattutto per l’aspetto facciale caratteristico, il rallentamento della crescita e le difficoltà nello sviluppo, ma può presentarsi in modi molto diversi da persona a persona. La maggior parte degli individui diagnosticati porta varianti in un gene chiave chiamato NIPBL, che aiuta a organizzare il modo in cui il DNA è impacchettato e letto all’interno delle cellule. Tuttavia, una porzione consistente di pazienti non presenta alterazioni in nessuno dei geni abituali, lasciando le famiglie senza risposte chiare. Poiché MAU2 lavora normalmente a stretto contatto con NIPBL nella stessa macchina cellulare, i ricercatori hanno ipotizzato che difetti sottili in MAU2 possano spiegare alcuni di questi casi inspiegati.
Collegare le varianti di MAU2 ai sintomi umani
I ricercatori hanno raccolto un gruppo di 18 individui portatori di varianti rare in una copia del gene MAU2. Queste varianti andavano da piccole sostituzioni di un singolo aminoacido a segnali di stop prematuro che troncano la proteina. Clinicamente, la maggior parte mostrava due tratti prominenti: bassa statura e microcefalia, cioè una dimensione della testa inferiore a quella attesa per l’età. Molti presentavano difficoltà di apprendimento da lievi a moderate e alcuni tratti facciali sovrapponibili alla sindrome di Cornelia de Lange, sebbene generalmente meno accentuati rispetto ai casi tipici correlati a NIPBL. Utilizzando un sistema di punteggio clinico consolidato per la sindrome, tre persone soddisfacevano i criteri per la forma “classica”, diverse rientravano in un intervallo “non classico” e alcuni erano così lievi da non essere probabilmente riconosciuti senza il test genetico.
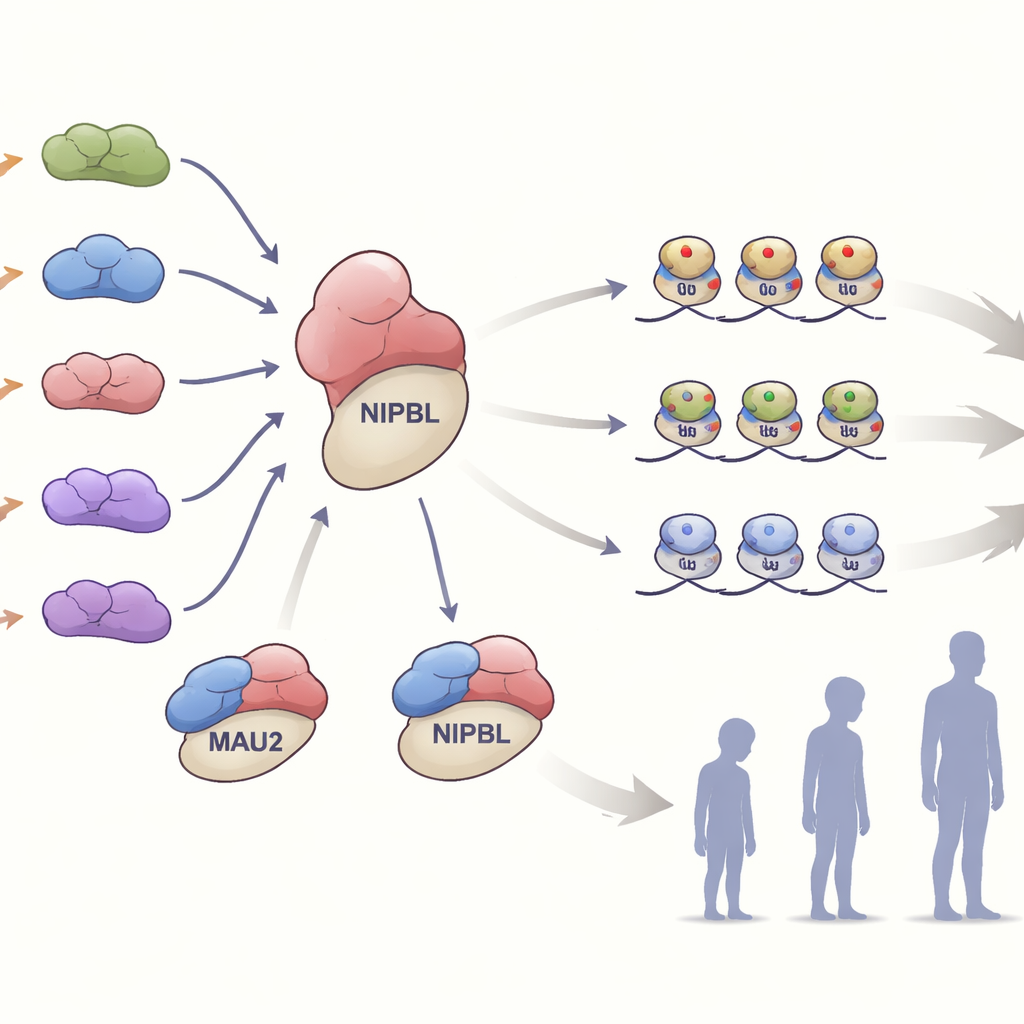
Come MAU2 difettoso altera l’organizzazione del genoma
All’interno delle cellule, MAU2 forma una stretta collaborazione con NIPBL per caricare sul DNA un complesso proteico ad anello chiamato cohesina. Questo complesso contribuisce a ripiegare il genoma e a modulare quali geni vengono attivati o disattivati. Il team ha testato come le diverse varianti di MAU2 influenzassero questa interazione. Molte delle varianti che rimuovevano o alteravano un tratto limitato della proteina ne indebolivano l’interazione con NIPBL senza distruggere nessuna delle due proteine. Altre varianti, che introdussero frameshift o segnali di stop anticipato, ridussero i livelli di MAU2 di circa la metà, una condizione nota come insufficienza di dose (haploinsufficienza). In cellule provenienti da una famiglia con una variante troncante, la riduzione della proteina MAU2 ha portato anche a una diminuzione dei livelli di NIPBL, sottolineando quanto i due partner dipendano l’uno dall’altro per la stabilità.
Leggere le impronte epigenetiche
Lo studio ha ampliato l’analisi oltre la sequenza del DNA esaminando le «impronte epigenetiche» sovrapposte al genoma. Nel sangue degli individui affetti, i ricercatori hanno misurato la metilazione del DNA, un marcatore chimico che spesso riflette come i geni sono regolati. La maggior parte dei portatori di varianti MAU2 mostrava un pattern di metilazione molto simile alla firma nota della sindrome di Cornelia de Lange, fornendo una solida evidenza molecolare che la perturbazione di MAU2 converge sulla stessa via patologica di NIPBL. In modo interessante, il team ha anche identificato due firme di metilazione specifiche per MAU2 che non si sovrapponevano completamente al pattern classico. Alcuni individui privi della consueta firma di Cornelia de Lange presentavano invece uno di questi profili specifici per MAU2 e avevano sintomi più lievi, suggerendo che una compromissione parziale di MAU2 lascia una propria traccia epigenetica riconoscibile.
Un modello murino riflette i cambiamenti umani nella crescita e nel cervello
Per valutare l’effetto della perdita di MAU2 sull’intero organismo, i ricercatori hanno studiato topi ingegnerizzati per avere una sola copia funzionante del gene Mau2, rispecchiando la situazione umana. I topi privi completamente di Mau2 non sopravvivevano, evidenziando l’essenzialità del gene. I topi con mezza dose sopravviventi erano leggermente più bassi dei fratelli normali e avevano cervelli più piccoli con regioni specifiche ridotte di dimensione, insieme a spazi liquidi ingranditi. Queste osservazioni riecheggiano la bassa statura, la microcefalia e le anomalie cerebrali sottili osservate nelle persone con varianti di MAU2, rafforzando il legame tra gene, sviluppo cerebrale e tratti phenotipici.
Cosa significa per le famiglie e per la diagnosi
Complessivamente, i dati umani e murini stabiliscono con forza MAU2 come gene capace di causare la sindrome di Cornelia de Lange e condizioni strettamente correlate. Le varianti che compromettono fortemente l’interazione MAU2–NIPBL tendono a produrre la firma epigenetica completa della sindrome e tratti facciali più riconoscibili, mentre varianti con effetti più lievi possono portare a bassa statura e microcefalia meno marcate con solo impronte molecolari specifiche per MAU2. Per le famiglie, questo significa che includere MAU2 nei pannelli diagnostici e considerare il profiling epigenetico può aiutare a spiegare casi precedentemente irrisolti, e mette in luce come anche piccole perturbazioni nella macchina che organizza il genoma possano riverberare fino a influenzare la crescita e lo sviluppo.
Citazione: Parenti, I., Hesters, A., Gil-Salvador, M. et al. Pathogenic variants in the cohesin loader subunit MAU2 underlie a distinct Cornelia de Lange Syndrome subtype. Nat Commun 17, 3036 (2026). https://doi.org/10.1038/s41467-026-71177-6
Parole chiave: sindrome di Cornelia de Lange, gene MAU2, loader della cohesina, metilazione del DNA, disturbi del neurosviluppo