Clear Sky Science · pt
Variantes patogênicas na subunidade carregadora de cohesina MAU2 estão na base de um subtipo distinto da síndrome de Cornelia de Lange
Quando uma Pequena Alteração Genética Molda Crescimento e Desenvolvimento
A síndrome de Cornelia de Lange é uma condição rara que afeta o crescimento, as feições faciais e a aprendizagem. Para muitas famílias, mesmo após testes genéticos, a causa continua desconhecida. Este estudo concentra-se em um gene menos estudado chamado MAU2 e mostra que alterações nesse gene podem causar uma forma distinta da síndrome de Cornelia de Lange, frequentemente associada a baixa estatura e microcefalia, mas com dificuldades de aprendizagem geralmente mais leves do que nos casos clássicos.
Uma Condição Rara com Muitas Aparências
A síndrome de Cornelia de Lange é mais conhecida por sua aparência facial característica, restrição do crescimento e desafios do desenvolvimento, mas pode se manifestar de formas muito diferentes de uma pessoa para outra. A maioria dos indivíduos diagnosticados carrega alterações em um gene-chave chamado NIPBL, que ajuda a organizar como o DNA é empacotado e lido dentro das células. Ainda assim, uma parcela considerável de pacientes não apresenta alterações em nenhum dos genes usuais, deixando as famílias sem respostas claras. Como o MAU2 normalmente funciona em conjunto com o NIPBL na mesma maquinaria celular, os pesquisadores suspeitaram que falhas sutis no MAU2 poderiam explicar alguns desses casos não esclarecidos.
Conectando Variantes de MAU2 aos Sintomas Humanos
Os pesquisadores reuniram um grupo de 18 indivíduos que apresentavam mudanças raras em uma cópia do gene MAU2. Essas alterações variaram desde pequenas trocas de um bloco construtor da proteína até sinais de parada prematuros que a truncam. Clinicamente, a maioria desses indivíduos compartilhava dois traços marcantes: baixa estatura e microcefalia, ou seja, tamanho da cabeça menor do que o esperado para a idade. Muitos tinham dificuldades de aprendizagem leves a moderadas e algumas feições faciais que se sobrepunham à síndrome de Cornelia de Lange, embora geralmente menos pronunciadas do que nos casos típicos relacionados ao NIPBL. Usando um sistema de pontuação clínica estabelecido para a síndrome, três pessoas se enquadraram nos critérios de Cornelia de Lange “clássica”, várias mais ficaram na faixa “não clássica” e algumas eram tão leves que provavelmente não teriam sido reconhecidas sem o teste genético.
Como o MAU2 Defeituoso Desorganiza o Genoma
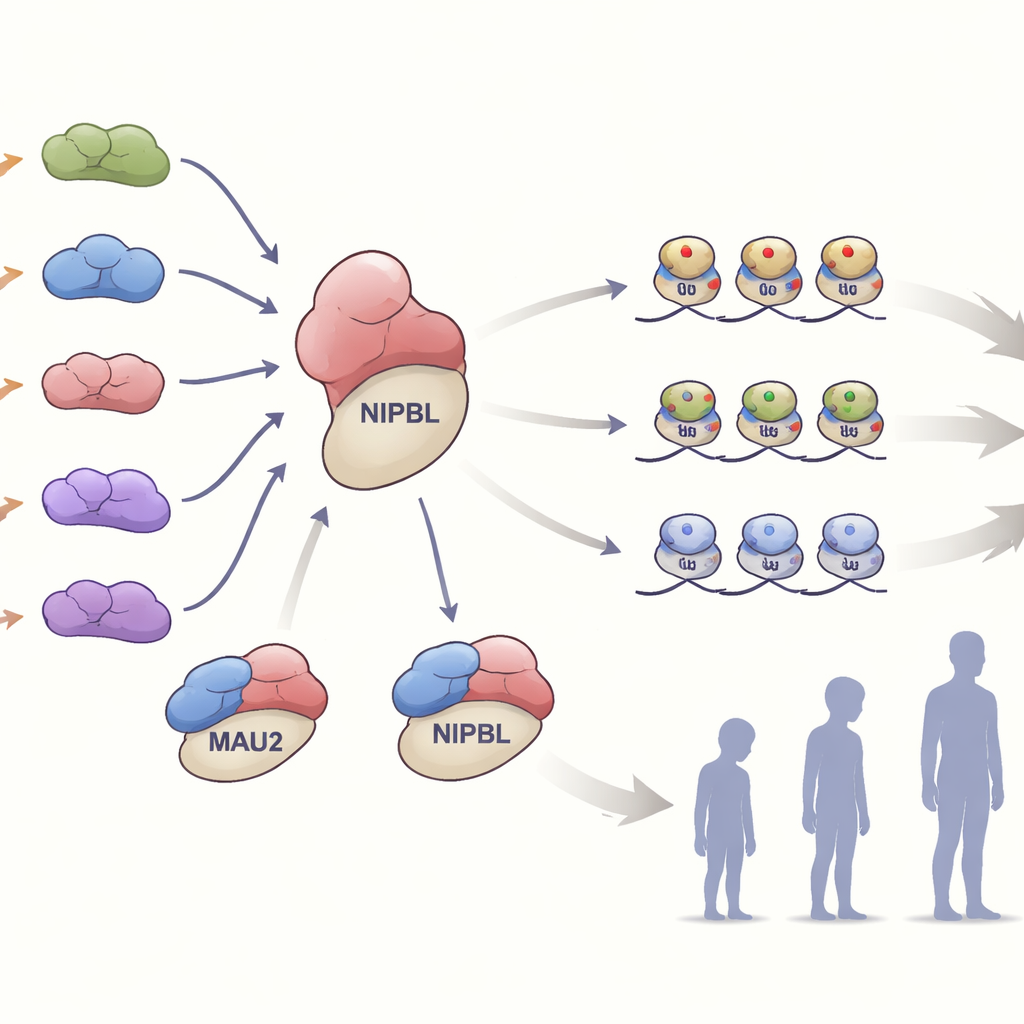
Dentro das células, o MAU2 forma uma parceria estreita com o NIPBL para carregar um complexo proteico em forma de anel chamado cohesina no DNA. Esse complexo ajuda a dobrar o genoma e a ajustar quais genes são ativados ou desativados. A equipe testou como as várias alterações em MAU2 afetavam essa parceria. Muitas das variantes que simplesmente removeram ou alteraram um pequeno trecho da proteína enfraqueceram sua interação com o NIPBL sem destruir nenhuma das proteínas por completo. Outras variantes, que introduziram quadros de leitura deslocados ou sinais de parada precoce, reduziram os níveis de MAU2 em cerca de metade, uma situação conhecida como haploinsuficiência. Em células de uma família com tal variante truncante, a queda na proteína MAU2 também reduziu os níveis de NIPBL, ressaltando o quão dependentes são esses dois parceiros para a estabilidade um do outro.
Lendo as Impressões Digitais Epigenéticas
O estudo foi além da sequência do DNA para examinar as marcas “epigenéticas” que se sobrepõem ao genoma. Em células sanguíneas dos indivíduos afetados, os pesquisadores mediram a metilação do DNA, uma marca química que frequentemente reflete como os genes são regulados. A maioria dos portadores de variantes em MAU2 mostrou um padrão de metilação que corresponde de perto à assinatura conhecida da síndrome de Cornelia de Lange, fornecendo forte evidência molecular de que a disrupção de MAU2 converge para a mesma via patológica do NIPBL. De modo intrigante, a equipe também descobriu duas assinaturas de metilação específicas de MAU2 que não se sobrepunham totalmente ao padrão clássico. Alguns indivíduos que não apresentavam a assinatura usual de Cornelia de Lange ainda carregavam um desses perfis específicos de MAU2 e tinham sintomas mais leves, sugerindo que a disrupção parcial de MAU2 pode deixar seu próprio traço epigenético reconhecível.
Um Modelo de Camundongo Reflete Mudanças em Crescimento e Cérebro Humanos
Para ver como a perda de MAU2 afeta um organismo inteiro, os pesquisadores estudaram camundongos engenheirados para portar apenas uma cópia funcional do gene Mau2, espelhando a situação humana. Camundongos totalmente isentos de Mau2 não sobreviveram, destacando a importância essencial do gene. Os animais com dose reduzida apresentaram estatura ligeiramente menor que a dos irmãos normais e cérebros menores com regiões específicas reduzidas em tamanho, além de espaços preenchidos por fluido aumentados. Essas descobertas ecoam a baixa estatura, a microcefalia e as anomalias cerebrais sutis observadas em pessoas com variantes em MAU2, fortalecendo a conexão entre gene, desenvolvimento cerebral e características externas.
O Que Isso Significa para Famílias e Diagnóstico
No conjunto, os dados humanos e de camundongo estabelecem de forma sólida o MAU2 como um gene que pode causar a síndrome de Cornelia de Lange e condições estreitamente relacionadas. Alterações que perturbam fortemente a parceria MAU2–NIPBL tendem a produzir a assinatura epigenética completa de Cornelia de Lange e feições faciais mais reconhecíveis, enquanto variantes com efeitos mais brandos podem levar a baixa estatura e microcefalia mais leves, com apenas impressões moleculares específicas de MAU2. Para as famílias, isso significa que adicionar o MAU2 aos painéis de testes diagnósticos e considerar o perfil epigenético pode ajudar a explicar casos previamente não resolvidos, e destaca como mesmo pequenas perturbações na maquinaria organizadora do genoma podem repercutir no crescimento e no desenvolvimento.
Citação: Parenti, I., Hesters, A., Gil-Salvador, M. et al. Pathogenic variants in the cohesin loader subunit MAU2 underlie a distinct Cornelia de Lange Syndrome subtype. Nat Commun 17, 3036 (2026). https://doi.org/10.1038/s41467-026-71177-6
Palavras-chave: síndrome de Cornelia de Lange, gene MAU2, carregador de cohesina, metilação do DNA, transtornos do desenvolvimento neurológico