Clear Sky Science · nl
Pathogene varianten in de cohesine-loader subeenheid MAU2 vormen een aparte subtype van het Cornelia de Lange-syndroom
Wanneer een kleine genetische verandering groei en ontwikkeling vormt
Het Cornelia de Lange-syndroom is een zeldzame aandoening die groei, gelaatstrekken en leren beïnvloedt. Voor veel gezinnen blijft de oorzaak, zelfs na genetisch onderzoek, onduidelijk. Deze studie richt zich op een minder bekend gen, MAU2, en toont aan dat veranderingen in dit gen een afzonderlijke vorm van het Cornelia de Lange-syndroom kunnen veroorzaken — vaak met kortere lichaamslengte en een kleine schedelomvang, maar over het algemeen mildere leerproblemen dan bij klassieke gevallen.
Een zeldzame aandoening met vele verschijningsvormen
Het Cornelia de Lange-syndroom staat vooral bekend om zijn karakteristieke gelaatstrekken, beperkte groei en ontwikkelingsproblemen, maar de uitingsvorm kan sterk variëren tussen personen. De meeste gediagnosticeerde individuen dragen veranderingen in een sleutelgen genaamd NIPBL, dat helpt bepalen hoe DNA in cellen wordt verpakt en afgelezen. Toch ontbreekt bij een substantieel deel van de patiënten een afwijking in de gebruikelijke genen, waardoor gezinnen zonder duidelijk antwoord blijven. Omdat MAU2 normaal gesproken nauw samenwerkt met NIPBL in dezelfde cellulaire machinerie, vermoedden onderzoekers dat subtiele fouten in MAU2 enkele van deze onverklaarde gevallen zouden kunnen verklaren.
MAU2-varianten koppelen aan menselijke symptomen
De onderzoekers verzamelden een groep van 18 personen die allemaal zeldzame veranderingen in één kopie van het MAU2-gen droegen. Deze veranderingen varieerden van kleine uitwisselingen van een enkel eiwitbouwblok tot voortijdige stopcodons die het eiwit afkappen. Klinisch deelden de meeste van deze personen twee opvallende kenmerken: een korte lichaamslengte en microcefalie, oftewel een kleinere schedelomvang dan verwacht voor de leeftijd. Velen hadden milde tot matige leerproblemen en sommige gelaatstrekken overlappenden met het Cornelia de Lange-syndroom, hoewel doorgaans minder uitgesproken dan bij typische NIPBL-gerelateerde gevallen. Met behulp van een vastgesteld klinisch scoresysteem voor het syndroom voldeden drie personen aan de criteria voor ‘klassiek’ Cornelia de Lange, vielen meerdere anderen in een ‘niet-klassiek’ bereik, en enkelen waren zo mild dat ze waarschijnlijk niet zouden zijn herkend zonder genetische tests.
Hoe foutief MAU2 de organisatie van het genoom verstoort
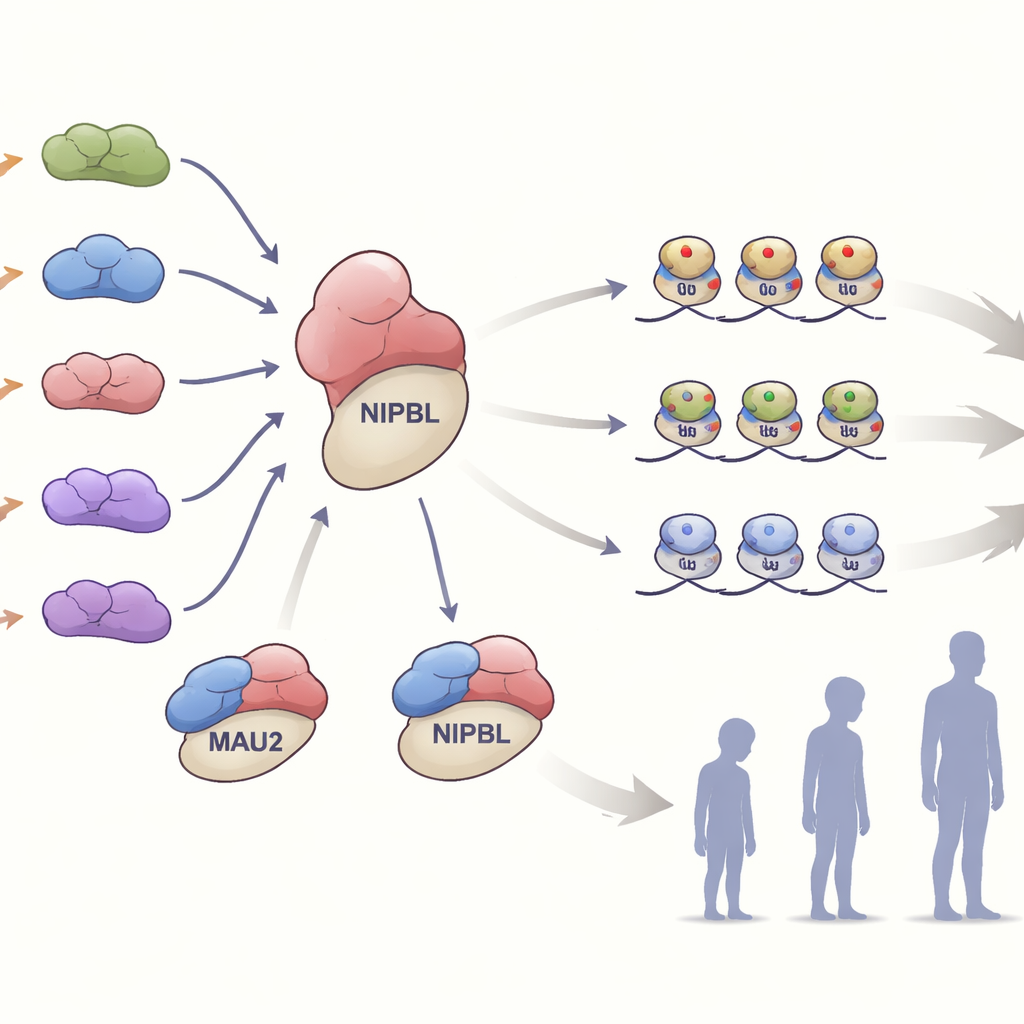
In cellen vormt MAU2 een nauwe samenwerking met NIPBL om een ringvormig eiwitcomplex, cohesine, op DNA te laden. Dit complex helpt het genoom op te vouwen en fijnmazig te regelen welke genen aan- of uitgezet worden. Het team onderzocht hoe de verschillende MAU2-veranderingen deze samenwerking beïnvloedden. Veel van de varianten die slechts een klein stukje van het eiwit verwijderden of veranderden, verzwakten de binding met NIPBL zonder een van beide eiwitten volledig te vernietigen. Andere varianten, die frameshifts of vroege stopcodons introduceerden, halveerden ruwweg de MAU2-niveaus — een situatie die bekendstaat als haploinsufficiëntie. In cellen van één familie met zo’n truncerende variant verminderde de daling van MAU2-eiwit ook de NIPBL-niveaus, wat benadrukt hoe sterk de stabiliteit van de twee partners van elkaar afhankelijk is.
De epigenetische vingerafdrukken lezen
De studie ging verder dan de DNA-sequentie en onderzocht de ‘epigenetische’ markeringen die bovenop het genoom liggen. In bloedcellen van aangetaste personen maten de onderzoekers DNA-methylering, een chemische markering die vaak weerspiegelt hoe genen worden gereguleerd. De meeste dragers van MAU2-varianten toonden een methylatiepatroon dat nauw overeenkwam met het bekende handtekeningprofiel van het Cornelia de Lange-syndroom, wat sterk moleculair bewijs levert dat verstoring van MAU2 convergeert op hetzelfde ziektepad als NIPBL. Intrigerend genoeg ontdekte het team ook twee MAU2-specifieke methylatiehandtekeningen die niet volledig overlappen met het klassieke patroon. Sommige personen die het gebruikelijke Cornelia de Lange-handtekening misten, droegen toch een van deze MAU2-specifieke profielen en hadden mildere symptomen, wat suggereert dat gedeeltelijke verstoring van MAU2 een eigen herkenbare epigenetische afdruk kan achterlaten.
Een muismodel weerspiegelt menselijke groei- en hersenveranderingen
Om te zien hoe verlies van MAU2 een heel organisme beïnvloedt, bestudeerden de onderzoekers muizen die zo waren gemodelleerd dat ze slechts één functionele kopie van het Mau2-gen hadden, wat de menselijke situatie nabootst. Muizen die volledig geen Mau2 hadden, overleefden niet, wat aantoont hoe essentieel het gen is. De overgebleven muizen met een halve dosis waren iets kleiner dan hun normale nestgenoten en hadden kleinere hersenen met specifieke regio’s die in omvang waren verminderd, samen met vergrote met vocht gevulde ruimtes. Deze bevindingen echoën de korte lichaamslengte, microcefalie en subtiele hersenanomalieën die bij mensen met MAU2-varianten worden waargenomen en versterken de link tussen gen, hersenontwikkeling en uiterlijke kenmerken.
Wat dit betekent voor gezinnen en diagnose
Gezamenlijk bevestigen de humane en muisgegevens dat MAU2 een gen is dat het Cornelia de Lange-syndroom en nauw verwante aandoeningen kan veroorzaken. Veranderingen die de MAU2–NIPBL-samenwerking sterk verstoren, neigen ertoe het volledige Cornelia de Lange-epigenetische handtekening en meer herkenbare gelaatstrekken te produceren, terwijl varianten met mildere effecten kunnen leiden tot een mildere korte lichaamslengte en kleine schedelomvang met alleen MAU2-specifieke moleculaire vingerafdrukken. Voor gezinnen betekent dit dat het opnemen van MAU2 in diagnostische panelen en het overwegen van epigenetische profilering kan helpen voorheen onopgeloste gevallen te verklaren, en het benadrukt hoe zelfs kleine verstoringen in de machinerie die het genoom organiseert, via kettingreacties groei en ontwikkeling kunnen beïnvloeden.
Bronvermelding: Parenti, I., Hesters, A., Gil-Salvador, M. et al. Pathogenic variants in the cohesin loader subunit MAU2 underlie a distinct Cornelia de Lange Syndrome subtype. Nat Commun 17, 3036 (2026). https://doi.org/10.1038/s41467-026-71177-6
Trefwoorden: Cornelia de Lange-syndroom, MAU2-gen, cohesine-loader, DNA-methylering, neuro-ontwikkelingsstoornissen