Clear Sky Science · pt
Neuroinflamação autoimune leva à morte neuronal via parthanatos mediado por nuclease MIF
Por que a inflamação no cérebro importa
A esclerose múltipla e doenças relacionadas não atacam apenas a bainha que isola as fibras nervosas—elas também matam lentamente os próprios neurônios. Essa perda oculta de neurônios impulsiona problemas de marcha, visão e cognição que podem continuar mesmo quando as recaídas estão controladas. O estudo resumido aqui pergunta algo básico, porém crucial: durante a inflamação autoimune no cérebro e na medula espinhal, como exatamente os neurônios morrem e é possível bloquear esse processo sem suprimir completamente o sistema imunológico?
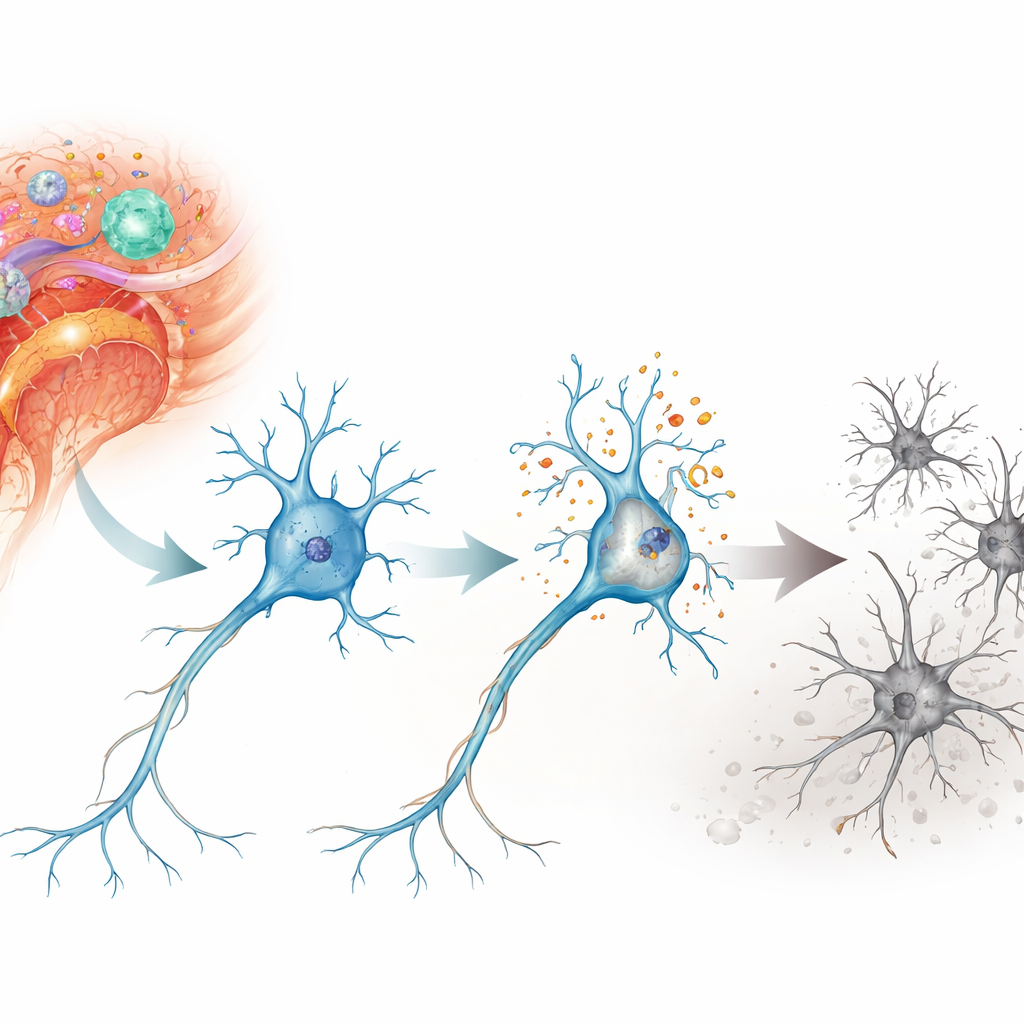
Uma reação em cadeia danosa dentro dos neurônios
Os pesquisadores focaram em uma forma de morte celular chamada parthanatos, um programa acionado por dano severo ao DNA, em contraste com a via mais conhecida do suicídio celular, a apoptose. Em um modelo murino de neuroinflamação autoimune conhecido como encefalomielite autoimune experimental, eles examinaram neurônios da medula espinhal e da retina—regiões lesionadas na esclerose múltipla. Encontraram fortes sinais de quebras de DNA e estresse oxidativo nesses neurônios, especialmente em neurônios motores da medula e em células ganglionares da retina que conectam o olho ao cérebro. Ao longo de dias a semanas, essas células foram progressivamente perdidas, acompanhando a evolução da incapacidade neurológica nos animais.
Seguindo um roteiro de morte chamado parthanatos
Observando com mais atenção, a equipe rastreou os passos moleculares do parthanatos dentro dos neurônios afetados. Quando o DNA era danificado, uma enzima nuclear chamada PARP1 tornava-se hiperativa e produzia longas cadeias de uma molécula chamada PAR. Essas cadeias vazavam para o corpo celular, onde ajudavam a desencadear a liberação de uma proteína mitocondrial (AIF) que se associa a outra proteína, o fator inibitório de migração de macrófagos (MIF). O complexo AIF–MIF então retornava ao núcleo. Lá, o MIF atuava como nuclease—uma enzima que corta DNA—causando fragmentação em larga escala do genoma e selando o destino do neurônio. Os autores detectaram cada uma dessas marcas, incluindo excesso de PAR, ligação AIF–MIF e acúmulo de MIF nos núcleos neuronais, durante o pico da doença nos camundongos e, em menor grau, em tecido cerebral humano de uma pessoa com esclerose múltipla.
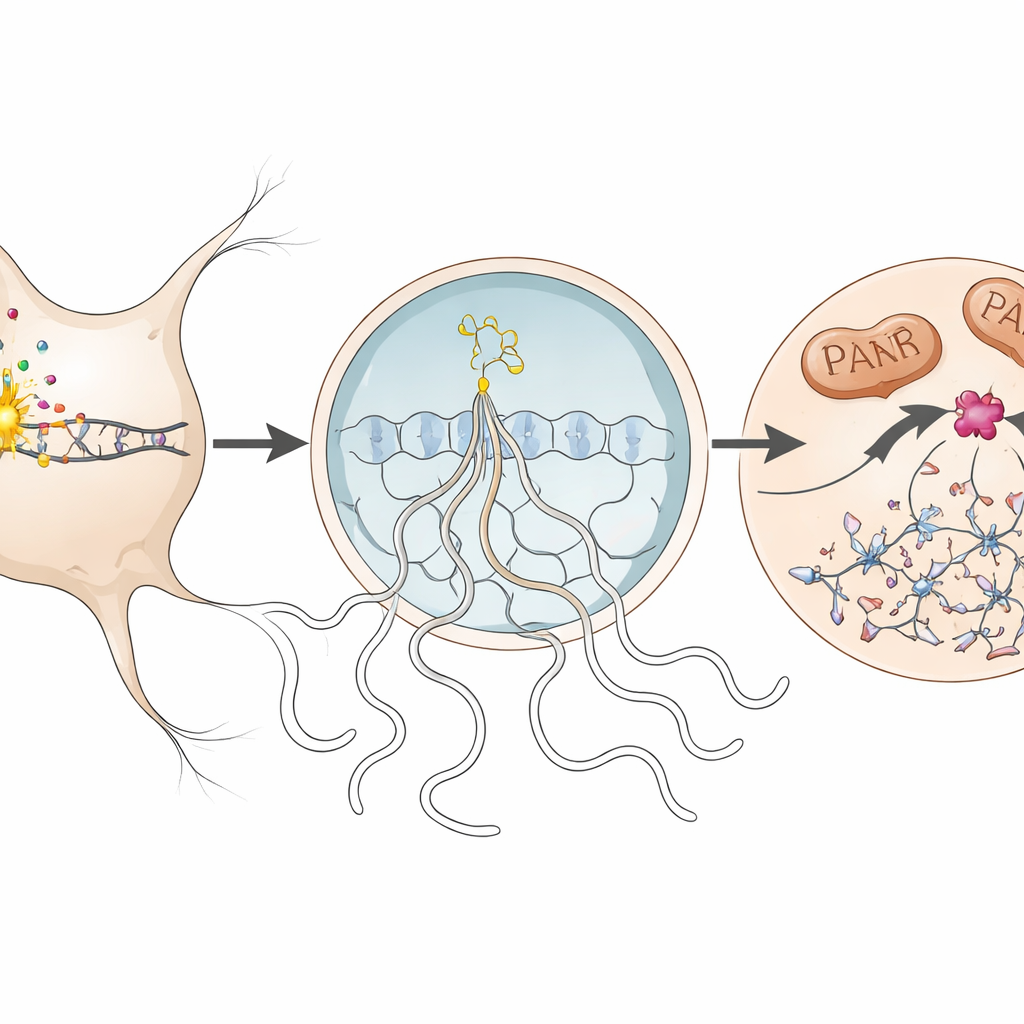
Protegendo neurônios ao desarmar uma única enzima
Para testar se essa via realmente impulsiona a perda neuronal, os investigadores desativaram a atividade de nuclease do MIF de duas maneiras complementares. Primeiro, usaram um camundongo knock-in que carrega uma mutação sutil em MIF (E22Q) que preserva suas outras funções, mas o impede de cortar DNA. Nesses animais, o ataque autoimune à mielina e a infiltração de células imunes na medula espinhal permaneceram inalterados, contudo os neurônios da medula e da retina foram significativamente mais preservados, e os escores de incapacidade a longo prazo foram menores do que em camundongos normais. Em segundo lugar, trataram os camundongos com PAANIB-1, uma pequena molécula que bloqueia seletivamente a atividade de nuclease do MIF. Seja administrado de forma preventiva ou após o início dos sintomas, esse fármaco reduziu a perda neuronal na fase crônica da doença sem alterar a infiltração de células imunes, a ativação glial ou o grau de desmielinização.
Neurônios respondem de modo diferente quando o parthanatos é bloqueado
A equipe então perguntou como os neurônios sobreviventes diferem ao nível da atividade gênica. Usando sequenciamento de RNA de núcleo único do tecido da medula espinhal, perfilaram dezenas de milhares de neurônios de camundongos normais e mutantes para MIF, com e sem doença. Em camundongos padrão com inflamação autoimune, os neurônios ativaram muitos genes relacionados ao sistema imune, incluindo vias que respondem a sinais de interferon e ajudam na apresentação de antígenos, enquanto reduziram genes importantes para a sinalização elétrica normal, comunicação por neurotransmissores e produção de fatores de crescimento protetores. Em contraste, neurônios de camundongos mutantes para MIF mantiveram grande parte dessa expressão gênica funcional central, mesmo que alguns programas inflamatórios tenham sido até mais fortemente ativados. Uma reanálise de dados cerebrais humanos com esclerose múltipla mostrou uma supressão semelhante e prolongada de genes básicos de função neuronal, sugerindo que os achados murinos refletem mudanças também em pessoas.
O que isso significa para tratamentos futuros
Em conjunto, os resultados apontam para o parthanatos—especificamente o passo final de corte do DNA levado a cabo pelo MIF—como uma via chave pela qual a inflamação mata neurônios em doenças autoimunes. Importante: bloquear a atividade de nuclease do MIF poupou neurônios sem atenuar a resposta imune mais ampla ou alterar a perda de mielina, e os neurônios protegidos pareceram preservar padrões mais saudáveis de atividade gênica. Para um leitor leigo, a conclusão principal é que este trabalho identifica um “interruptor” molecular concreto para um programa destrutivo de morte dentro dos neurônios. Fármacos direcionados que acionem esse interruptor poderiam, em princípio, acrescentar verdadeira neuroproteção às terapias já focadas no sistema imune para esclerose múltipla e outras condições inflamatórias do cérebro e da medula espinhal.
Citação: Mace, J.W., Gadani, S.P., Smith, M.D. et al. Autoimmune neuroinflammation leads to neuronal death via MIF nuclease-mediated parthanatos. Nat Neurosci 29, 796–809 (2026). https://doi.org/10.1038/s41593-026-02201-7
Palavras-chave: esclerose múltipla, neuroinflamação, morte de células neuronais, parthanatos, nuclease MIF