Clear Sky Science · pl
Autoimmunologiczne zapalenie mózgu prowadzi do śmierci neuronów przez parthanatos zależny od nukleazy MIF
Dlaczego zapalenie w mózgu ma znaczenie
Stwardnienie rozsiane i choroby pokrewne nie tylko atakują osłonkę włókien nerwowych — stopniowo zabijają także same neurony. To ukryte ubytki neuronów napędzają problemy z chodzeniem, wzrokiem i funkcjami poznawczymi, które mogą postępować nawet wtedy, gdy nawroty choroby są kontrolowane. Badanie opisane tutaj stawia fundamentalne, ale istotne pytanie: w trakcie autoimmunologicznego zapalenia mózgu i rdzenia kręgowego, w jaki sposób dokładnie giną neurony i czy można zablokować ten proces bez całkowitego tłumienia układu odpornościowego?
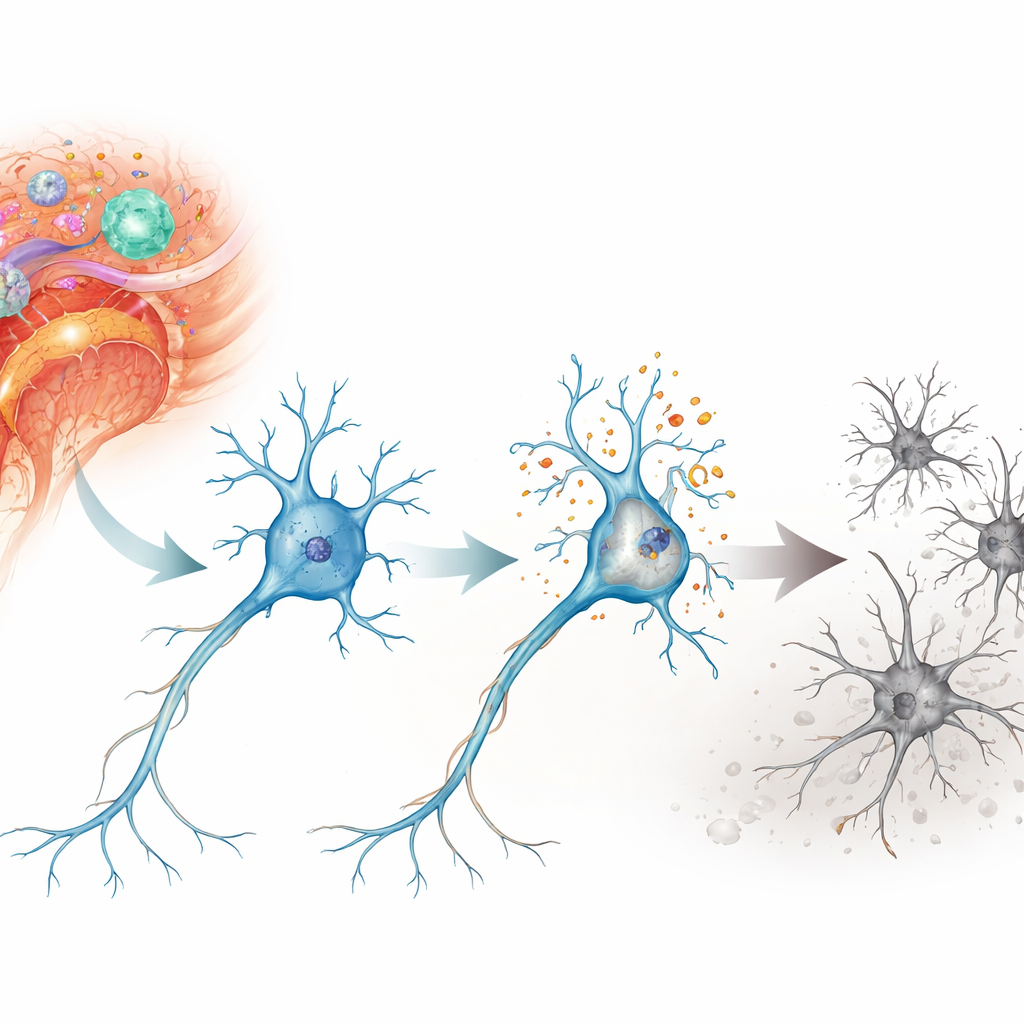
Szkodliwy łańcuch reakcji w neuronach
Naukowcy skupili się na formie śmierci komórkowej zwanej parthanatosem — programie uruchamianym przez poważne uszkodzenia DNA, a nie przez bardziej znaną ścieżkę samobójczą, czyli apoptozę. W modelu myszy z autoimmunologicznym zapaleniem układu nerwowego znanym jako eksperymentalne autoimmunologiczne zapalenie mózgu i rdzenia badali neurony w rdzeniu kręgowym i siatkówce — obszarach uszkadzanych w stwardnieniu rozsianym. Stwierdzili silne oznaki pęknięć DNA i stresu oksydacyjnego w tych neuronach, szczególnie w neuronach ruchowych rdzenia kręgowego oraz komórkach zwojowych siatkówki łączących oko z mózgiem. W ciągu dni do tygodni komórki te stopniowo ulegały utracie, co odpowiadało czasowi narastania upośledzenia neurologicznego u zwierząt.
Śledząc program śmierci zwany parthanatosem
Przy bliższym przyjrzeniu się zespół odtworzył molekularne kroki parthanatosu w dotkniętych neuronach. Gdy DNA było uszkodzone, enzym jądrowy PARP1 ulegał nadaktywacji i wytwarzał długie łańcuchy cząsteczki PAR. Te łańcuchy przedostawały się do ciała komórkowego, gdzie pomagały wywołać uwolnienie białka mitochondrialnego (AIF), które współdziałało z innym białkiem — inhibitorem migracji makrofagów (MIF). Kompleks AIF–MIF następnie przemieszczał się z powrotem do jądra komórkowego. Tam MIF działał jako nukleaza — enzym przecinający DNA — powodując masową fragmentację genomu i przypieczętowując los neuronu. Autorzy wykryli każdy z tych znaków: nadmiar PAR, wiązanie AIF–MIF oraz akumulację MIF w jądrach neuronów w szczytowej fazie choroby u myszy, a w mniejszym zakresie także w tkance mózgowej od osoby ze stwardnieniem rozsianym.
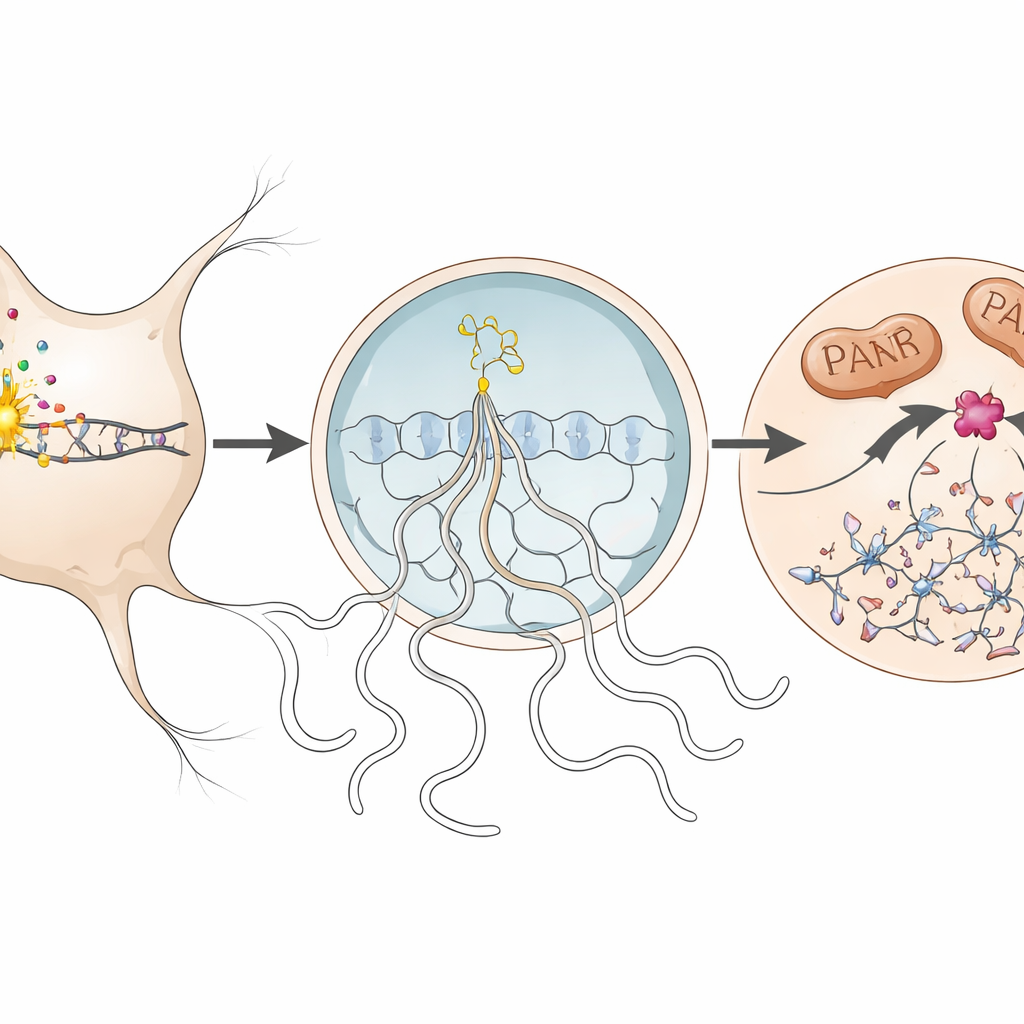
Ochrona neuronów przez unieszkodliwienie jednego enzymu
Aby sprawdzić, czy ta ścieżka rzeczywiście odpowiada za utratę neuronów, badacze zablokowali aktywność nukleazową MIF na dwa komplementarne sposoby. Po pierwsze użyli myszy z wprowadzonym miejscowym wariantem MIF (E22Q), który pozostawia inne funkcje białka nienaruszone, ale uniemożliwia mu cięcie DNA. U tych zwierząt autoimmunologiczny atak na osłonkę mielinową i napływ komórek odpornościowych do rdzenia kręgowego pozostały niezmienione, jednak neurony w rdzeniu i siatkówce były istotnie lepiej zachowane, a długoterminowe wyniki sprawności były niższe niż u myszy normalnych. Po drugie, leczono myszy związkiem PAANIB-1, małą cząsteczką selektywnie blokującą aktywność nukleazową MIF. Zarówno podawany profilaktycznie, jak i po wystąpieniu objawów, lek zmniejszał utratę neuronów w przewlekłej fazie choroby, nie wpływając na infiltrację komórek odpornościowych, aktywację gleju ani stopień demielinizacji.
Neurony reagują inaczej, gdy parthanatos jest zablokowany
Zespół zapytał następnie, czym różnią się przeżywające neurony na poziomie aktywności genów. Wykorzystując sekwencjonowanie RNA pojedynczych jąder z tkanki rdzenia kręgowego, przeanalizowali dziesiątki tysięcy neuronów z myszy normalnych i z mutantem MIF, z chorobą i bez niej. U standardowych myszy z autoimmunologicznym zapaleniem neurony włączały wiele genów związanych z odpowiedzią immunologiczną, w tym szlaki reagujące na sygnały interferonowe i uczestniczące w prezentacji antygenów, przy jednoczesnym wyciszeniu genów ważnych dla normalnego przewodzenia elektrycznego, komunikacji neuroprzekaźnikowej i produkcji ochronnych czynników wzrostu. Natomiast neurony u myszy z mutacją MIF zachowały znaczną część tego podstawowego programu genów funkcjonalnych, chociaż niektóre programy zapalne były nawet silniej aktywowane. Ponowna analiza danych z mózgów ludzi ze stwardnieniem rozsianym ujawniła podobne długotrwałe tłumienie podstawowych genów funkcji neuronalnych, co sugeruje, że wyniki u myszy odzwierciedlają zmiany u ludzi.
Co to oznacza dla przyszłych terapii
Podsumowując, wyniki wskazują na parthanatos — a w szczególności na końcowy krok cięcia DNA przeprowadzany przez MIF — jako kluczową drogę, przez którą zapalenie zabija neurony w chorobach autoimmunologicznych. Co ważne, blokowanie aktywności nukleazowej MIF chroniło neurony bez tłumienia szerokiej odpowiedzi immunologicznej ani zmiany utraty mieliny, a chronione neurony wykazywały zdrowsze wzorce ekspresji genów. Dla czytelnika niebędącego specjalistą sedno jest takie: praca ta identyfikuje konkretny molekularny „wyłącznik” destrukcyjnego programu śmierci w neuronach. Leki celowane na ten wyłącznik mogłyby w zasadzie dodać rzeczywistą neuroprotekcję do istniejących terapii ukierunkowanych na układ odpornościowy przy stwardnieniu rozsianym i innych zapalnych schorzeniach mózgu oraz rdzenia kręgowego.
Cytowanie: Mace, J.W., Gadani, S.P., Smith, M.D. et al. Autoimmune neuroinflammation leads to neuronal death via MIF nuclease-mediated parthanatos. Nat Neurosci 29, 796–809 (2026). https://doi.org/10.1038/s41593-026-02201-7
Słowa kluczowe: stwardnienie rozsiane, nezapalenie układu nerwowego, obumieranie neuronów, parthanatos, nukleaza MIF