Clear Sky Science · pl
Rzadkie choroby genetyczne związane ze stresem replikuacyjnym wywołanym przez G-kwadrupleksy
Kiedy supły DNA prowadzą do rzadkich chorób
Nasz materiał genetyczny zwykle wyobrażany jest jako gładka, skręcona drabina. Jednak w wielu miejscach DNA może składać się w zwarte supły zwane G‑kwadrupleksami. Te nietypowe struktury występują szczególnie często w ważnych regionach kontrolnych genomu i w pobliżu końców chromosomów. Ten przegląd wyjaśnia, jak takie supły mogą spowalniać lub zatrzymywać kopiowanie DNA, tworząc „stres replikuacyjny”, który uszkadza chromosomy i przyczynia się do zaskakująco szerokiego spektrum rzadkich, dziedzicznych chorób, często związanych z zaburzeniami wzrostu, defektami odporności, przedwczesnym starzeniem się i ryzykiem nowotworów.
Nietypowe kształty DNA w naszym genomie
G‑kwadrupleksy tworzą się w odcinkach DNA bogatych w literę G (guanina). Zamiast parować w zwykły sposób, cztery guaniny mogą układać się w płaskie „płytki”, które układają się jedna na drugiej tworząc stabilny słupek. Nowoczesne techniki mapowania wykrywają setki tysięcy takich potencjalnych miejsc rozrzuconych po ludzkim genomie, szczególnie w przełącznikach genów (promotorach), niekodujących regionach RNA oraz ochronnych czapkach chromosomów zwanych telomerami. W komórkach występują też białka rozpoznające lub stabilizujące te struktury. Wspólnie sugeruje to, że G‑kwadrupleksy nie są rzadkimi osobliwościami, lecz powtarzającymi się elementami wpływającymi na to, jak geny są włączane, jak rozmieszczane są chemiczne modyfikacje DNA i jak utrzymywane są telomery.
Kiedy kopiowanie DNA napotyka przeszkodę
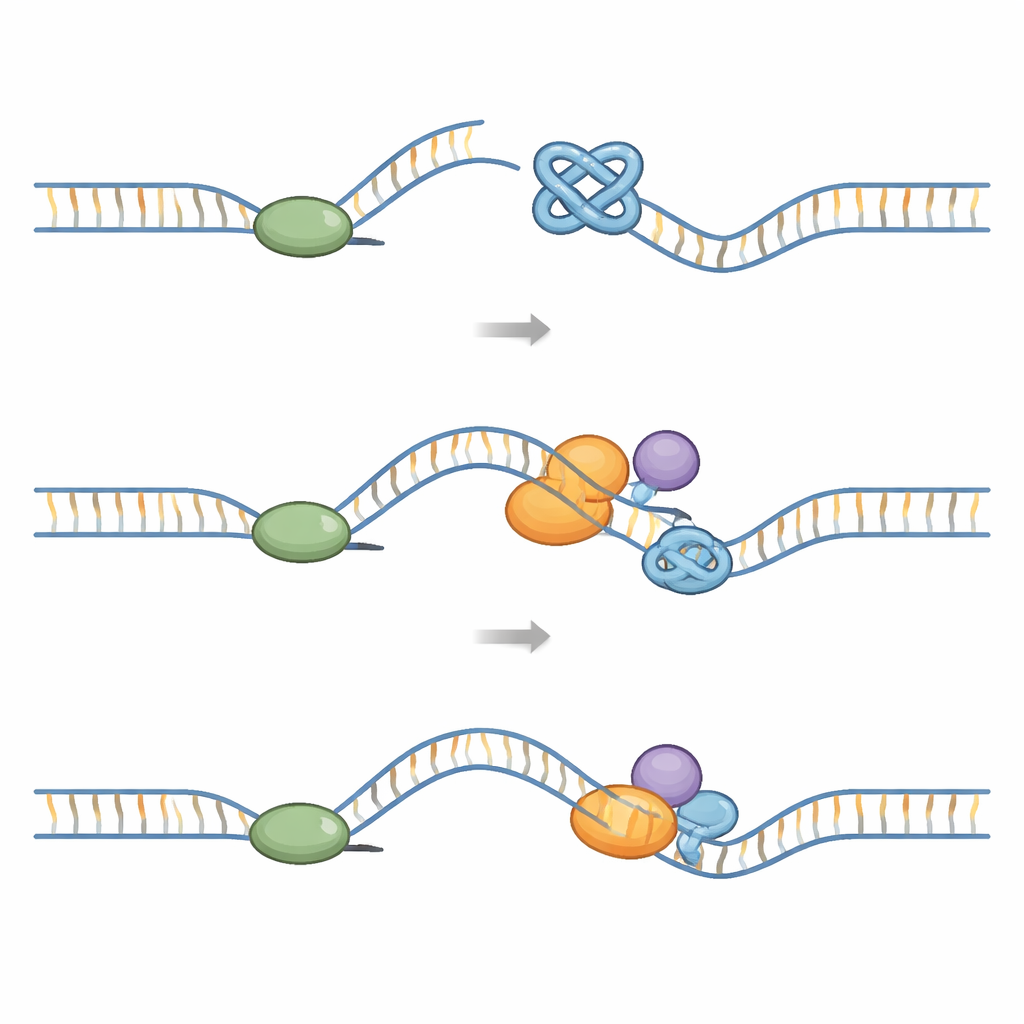
Za każdym razem, gdy komórka się dzieli, duży kompleks „replisomu” musi zduplikować cały genom. W skład tego mechanizmu wchodzi helikaza w kształcie pierścienia, która rozdziela DNA, oraz polimerazy budujące nowe nici. G‑kwadrupleksy stanowią mechaniczne wyzwanie: są trudniejsze do rozdzielenia niż zwykłe DNA. Badania strukturalne pokazują, że gdy replisom napotyka G‑kwadrupleks, supły mogą zaklinować się wewnątrz pierścienia helikazy i zablokować jej ruch. Inne eksperymenty wykazują, że G‑kwadrupleksy utworzone na odsłoniętej nici matrycowej mogą zatrzymać samą polimerazę i spowodować rozłączenie helikazy i polimerazy. Jeśli takie zatory nie zostaną usunięte, widełki repliksacyjne mogą się złamać, prowadząc do pęknięć podwójnych nici DNA, rearranżacji chromosomowych i miejsc kruchego DNA, szczególnie na telomerach.
Specjalistyczne enzymy rozwiązujące supły G‑kwadrupleksów
Komórki polegają na sieci „pomocniczych” enzymów, aby zapobiec trwałym szkodom powodowanym przez G‑kwadrupleksy. Kilka rodzin helikaz — małych silniczków poruszających się po DNA i rozwijających je — odgrywa tu szczególną rolę. Helikazy z rodziny RecQ, takie jak BLM, WRN, RECQL4 i RECQL1, potrafią rozpoznawać i rozplątywać G‑kwadrupleksy, także te na telomerach. Druga grupa, helikazy zawierające centra żelazo‑siarka (Fe–S) jak FANCJ, RTEL1 i DDX11, oraz enzymy takie jak DNA2 i PIF1, również rozkładają G‑kwadrupleksy lub je przecinają. Niektóre z nich wykształciły specjalne kieszenie lub kliny chwytające pojedyncze guaniny lub konkretne kształty G‑kwadrupleksów, co pozwala im stopniowo rozdzielać te supły, współpracując z białkami wiążącymi DNA i czynnikami replikacyjnymi. Inne przebudowujące białka, takie jak translokaza DNA HLTF i helikaza G‑kwadrupleksów/R‑pętli DHX36, przekształcają zablokowane widełki lub pomagają replisomowi ominąć przeszkody, aby kopiowanie mogło zostać ukończone.
Jak wadliwe „pomocniki” DNA powodują rzadkie zaburzenia
Ponieważ białka radzące sobie z G‑kwadrupleksami pełnią też szersze role w naprawie i odpowiedzi na stres, dziedziczne mutacje w ich genach prowadzą do charakterystycznych rzadkich chorób. Defekty w BLM powodują zespół Blooma, charakteryzujący się zahamowaniem wzrostu, problemami odpornościowymi i wysokim ryzykiem nowotworów. Mutacje w WRN prowadzą do zespołu Wernera, formy przedwczesnego starzenia z wczesną zaćmą, cukrzycą i utratą masy kostnej. Zmiany w RTEL1 leżą u podstaw zaburzeń związanych z telomerami, takich jak dyskeratoza wrodzona i zespół Hoyeraal–Hreidarssona, które obejmują niewydolność szpiku i bardzo krótkie telomery. Mutacje w FANCJ i BRCA2 wiążą się z anemią Fanconiego i silną predyspozycją do nowotworów, podczas gdy mutacje w DDX11 powodują Zespół Warszawskiego Łamania z mikrocefalią i defektami kohezji sióstr chromosomów. W wielu z tych schorzeń komórki wykazują nagromadzenie G‑kwadrupleksów, zatrzymane widełki replikacyjne, kruche telomery i pęknięcia chromosomowe — wszystkie cechy stresu replikuacyjnego wywołanego przez G‑kwadrupleksy.
Od rzadkich zaburzeń genetycznych do nowych terapii
Autorzy twierdzą, że G‑kwadrupleksy znajdują się na skrzyżowaniu struktury DNA, replikacji i choroby. Gdy są właściwie kontrolowane, mogą pomagać w regulacji genów i telomerów; gdy nie, stają się przeszkodami uszkadzającymi genom i przyczyniają się do rzadkich zespołów, neurodegeneracji i możliwie niektórych aspektów starzenia. Dokładne poznanie, jak różne enzymy rozpoznają i rozplątują konkretne kształty G‑kwadrupleksów, daje dwie możliwości: poprawę diagnostyki rzadkich zaburzeń związanych ze stresem replikuacyjnym oraz projektowanie ukierunkowanych terapii. Z jednej strony są leki stabilizujące G‑kwadrupleksy, mające selektywnie zabijać komórki nowotworowe o osłabionych systemach naprawczych, co sugerują wstępne badania kliniczne. Z drugiej pojawiają się strategie — takie jak białka kierowane przez CRISPR — które kiedyś mogłyby modulować tworzenie G‑kwadrupleksów w wybranych miejscach, aby skorygować zaburzenia kontroli genów. W obu przypadkach postrzeganie DNA nie tylko jako podwójnej helisy, lecz jako krajobrazu dynamicznych struktur jest kluczem do zrozumienia, a ostatecznie leczenia, tych chorób.
Cytowanie: Herr, L.M., Mukhopadhyay, S., Anderson, O.M. et al. Rare genetic diseases associated with G-quadruplex-induced replication stress. Commun Biol 9, 522 (2026). https://doi.org/10.1038/s42003-026-09966-4
Słowa kluczowe: DNA G-kwadrupleksy, stres replikuacyjny, rzadkie choroby genetyczne, helikazy DNA, niestabilność telomerów