Clear Sky Science · pl
Syntaza aminoadypinian‑półaldehydu — potencjalny cel terapii redukcji substratu w kwasicy glutarynowej typu 1
Dlaczego ta rzadka choroba dziecięca ma znaczenie
Kwasica glutarynowa typu 1 to rzadka choroba dziedziczna, która przede wszystkim dotyka niemowlęta i małe dzieci, ale stanowi ilustrację znacznie szerszej idei: jak modyfikacja jednego kroku w biochemii organizmu może chronić mózg przed trwałymi uszkodzeniami. Pomimo współczesnego przesiewu noworodków i ostrożnych diet, wiele dzieci z tą chorobą i tak rozwija zaburzenia ruchowe, zmiany w mózgu oraz problemy z nerkami. W badaniu tym analizuje się nową metodę ograniczenia powstawania szkodliwych substancji u źródła, zwaną terapią redukcji substratu, i sprawdza się, czy może ona doprowadzić do bezpieczniejszych i skuteczniejszych terapii.
Korek w szlakach metabolicznych organizmu
W kwasicy glutarynowej typu 1 organizm nie potrafi prawidłowo rozkładać niektórych składników białek, szczególnie aminokwasu lizyny. Brak lub nieprawidłowość enzymu zwanego dehydrogenazą glutarylo‑CoA powoduje metaboliczny korek, prowadząc do gromadzenia się kwasu glutarynowego i powiązanych związków. Substancje te są szczególnie toksyczne dla określonych rejonów mózgu kontrolujących ruch, zwłaszcza we wczesnym okresie życia. Nawet gdy dzieci zostają zidentyfikowane krótko po narodzinach i wprowadza się u nich specjalne diety ubogie w lizynę, około jedna trzecia nadal rozwija poważne objawy neurologiczne, a wiele z nich z czasem wykazuje postępujące zmiany istoty białej i problemy nerkowe.
Nowe miejsce do postawienia blokady
Obecne leczenie skupia się na ograniczeniu ilości lizyny dostającej się do organizmu i wspomaganiu usuwania toksycznych produktów ubocznych, ale nie zmienia bezpośrednio działania wadliwego szlaku. Badacze postawili kluczowe pytanie: zamiast radzić sobie z nagromadzonymi odpadami, czy można spowolnić dopływ substratów do wadliwego szlaku? Skoncentrowali się na innym enzymie — syntazie aminoadypinian‑półaldehydu (AASS), która przeprowadza pierwsze dwa etapy jednego z głównych szlaków rozkładu lizyny. Osoby naturalnie pozbawione tego enzymu zazwyczaj mają wysokie stężenia lizyny, ale niewiele lub żadnych problemów zdrowotnych, co sugeruje, że jego zablokowanie może być stosunkowo bezpieczne.
Testy pomysłu na zmodyfikowanych myszach
Aby zbadać tę strategię, zespół użył myszy hodowanych bez dehydrogenazy glutarylo‑CoA, dobrze znanego modelu tej choroby u ludzi. Zwierzęta te gromadzą duże ilości toksycznych kwasów w tkankach i po podaniu diety bogatej w lizynę rozwijają drgawki, problemy ruchowe, uszkodzenia mózgu, a często też umierają. Badacze stworzyli następnie mysz z podwójnym „wyłączeniem” — pozbawioną zarówno enzymu chorobowego, jak i syntazy aminoadypinian‑półaldehydu. Przy standardowym podejściu żywieniowym myszy z podwójnym knockoutem wyglądały i zachowywały się jak zdrowe zwierzęta w szerokim zestawie testów, wykazując jedynie łagodne nieprawidłowości biochemiczne.
Mniej toksycznego nagromadzenia i lepsze zdrowie mózgu
Prawdziwy test przyszedł, gdy oba typy myszy‑modeli choroby zostały wystawione na dietę wysoką w lizynę, zaprojektowaną tak, by obciążyć wrażliwy szlak. Myszy pozbawione tylko enzymu chorobowego akumulowały bardzo wysokie poziomy toksycznych kwasów w mózgu, wątrobie, nerkach, krwi i moczu, a wiele z nich rozwijało ciężkie objawy neurologiczne, drgawki i utratę masy ciała. Dla porównania, zwierzęta z podwójnym wyłączeniem miały dramatycznie niższe stężenia kwasu glutarynowego we wszystkich tych tkankach, znacznie mniej oznak choroby i zachowanie zbliżone do normalnego. Mikroskopowe badanie mózgów wykazało, że drobne wakuole i zmiany strukturalne obecne u myszy z pojedynczym knockoutem były znacznie zredukowane lub nieobecne u myszy z podwójnym knockoutem. Co ważne, szczegółowe analizy składu ciała, gęstości kości, funkcji serca, markerów odpornościowych i chemii krwi nie wykazały istotnych pozanerwowych problemów spowodowanych zablokowaniem tego wczesnego kroku rozkładu lizyny.
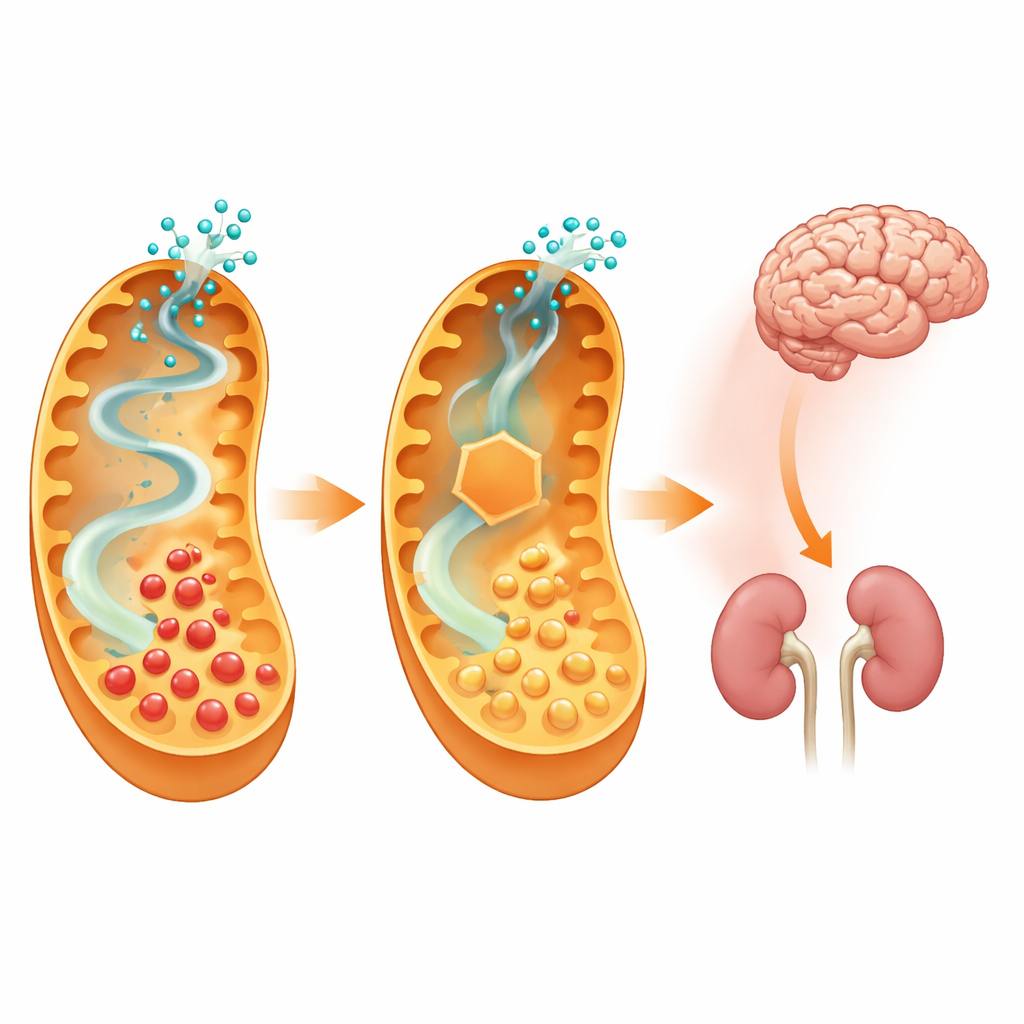
Co to oznacza dla przyszłych terapii
Wyłączając pierwszy etap konkretnego szlaku rozkładu lizyny, badacze byli w stanie zmniejszyć narażenie mózgu na toksyczne metabolity i w dużym stopniu ochronić myszy przed ciężkim, wywołanym dietą zaostrzeniem choroby, bez wywoływania widocznych nowych problemów zdrowotnych. Sugeruje to, że leki zaprojektowane do częściowej inhibicji syntazy aminoadypinian‑półaldehydu mogłyby stanowić potężne uzupełnienie diety i innych terapii dla dzieci z kwasicą glutarynową typu 1. Chociaż obecne badanie opiera się na inżynierii genetycznej u myszy, a odpowiednie leki nadal muszą zostać opracowane i przebadane pod kątem bezpieczeństwa u ludzi, praca ta dostarcza silnego dowodu koncepcji, że celowane zablokowanie wstępnych etapów chemii metabolicznej może zmienić przebieg wyniszczającej metabolicznej choroby mózgu.
Cytowanie: Saad, C., Jung-Klawitter, S., Dimitrov, B. et al. Aminoadipate-semialdehyde synthase, a potential target for substrate reduction therapy in glutaric aciduria type 1. Sci Rep 16, 10995 (2026). https://doi.org/10.1038/s41598-026-44377-9
Słowa kluczowe: kwasica glutarynowa typu 1, metabolizm lizyny, terapia redukcji substratu, inhibicja AASS, dziedziczne choroby metaboliczne