Clear Sky Science · fr
Synthase de l’aminoadipate-semialdéhyde, une cible potentielle pour une thérapie de réduction du substrat dans l’acidurie glutarique de type 1
Pourquoi cette maladie infantile rare compte
L’acidurie glutarique de type 1 est une maladie héréditaire rare qui touche principalement les nourrissons et les jeunes enfants, mais elle illustre une idée beaucoup plus large : comment modifier une seule étape de la chimie corporelle peut protéger le cerveau contre des lésions à vie. Malgré le dépistage néonatal moderne et des régimes alimentaires contrôlés, de nombreux enfants atteints développent encore des troubles du mouvement, des altérations cérébrales et des problèmes rénaux. Cette étude explore une nouvelle façon de couper à la source la production de substances nocives, en utilisant une stratégie appelée thérapie de réduction du substrat, et teste si cela pourrait conduire à des traitements plus sûrs et plus efficaces.
Un embouteillage dans la chimie du corps
Dans l’acidurie glutarique de type 1, l’organisme ne peut pas décomposer correctement certains éléments constitutifs des protéines, en particulier l’acide aminé lysine. Une enzyme manquante ou défectueuse, connue sous le nom de glutaryl-CoA déshydrogénase, provoque un embouteillage métabolique, entraînant l’accumulation d’acide glutarique et de composés apparentés. Ces substances sont particulièrement toxiques pour des régions cérébrales spécifiques impliquées dans le contrôle du mouvement, surtout tôt dans la vie. Même lorsque les enfants sont identifiés peu après la naissance et placés sous régimes spéciaux pauvres en lysine, environ un sur trois développe encore des symptômes neurologiques graves, et beaucoup présentent à mesure qu’ils grandissent une atteinte progressive de la matière blanche et des problèmes rénaux.
Un nouvel endroit où placer un obstacle
Le traitement actuel vise à limiter la quantité de lysine qui entre dans l’organisme et à aider à éliminer les sous-produits toxiques, mais il ne modifie pas directement le fonctionnement de la voie défectueuse. Les chercheurs ont posé une question clé : au lieu d’essayer de gérer les déchets qui s’accumulent, pourrait-on ralentir l’entrée dans la voie défectueuse elle‑même ? Ils se sont concentrés sur une autre enzyme appelée synthase de l’aminoadipate‑semialdéhyde, qui réalise les deux premières étapes d’une des principales voies de dégradation de la lysine. Les personnes qui manquent naturellement cette enzyme présentent généralement des taux élevés de lysine mais peu ou pas de problèmes de santé, ce qui suggère que la bloquer pourrait être relativement sûr.
Tester l’idée chez des souris génétiquement modifiées
Pour explorer cette stratégie, l’équipe a utilisé des souris élevées sans glutaryl‑CoA déshydrogénase, un modèle bien établi de la maladie humaine. Ces animaux accumulent de grandes quantités d’acides toxiques dans leurs tissus et, lorsqu’ils sont soumis à un régime riche en lysine, développent des convulsions, des troubles du mouvement, des lésions cérébrales et meurent souvent. Les chercheurs ont ensuite créé une souris à double knockout, dépourvue à la fois de l’enzyme de la maladie et de la synthase de l’aminoadipate‑semialdéhyde. Dans des conditions d’alimentation standard, ces souris à double knockout paraissaient et se comportaient comme des animaux sains dans une large batterie de tests, tout en ne présentant que de légères anomalies biochimiques.
Moins d’accumulation toxique et une meilleure santé cérébrale
Le véritable test a eu lieu lorsque les deux types de souris modèles de la maladie ont été exposés à un régime riche en lysine conçu pour solliciter la voie vulnérable. Les souris ne présentant que l’absence de l’enzyme de la maladie accumulaient des niveaux très élevés d’acides toxiques dans le cerveau, le foie, les reins, le sang et l’urine, et nombre d’entre elles développaient de sévères symptômes neurologiques, des convulsions et une perte de poids. En revanche, les animaux à double knockout avaient des taux d’acide glutarique considérablement plus faibles dans tous ces tissus, beaucoup moins de signes de maladie et un comportement presque normal. L’examen microscopique de leurs cerveaux a montré que les petites vacuoles et les modifications structurelles observées chez les souris à knockout simple étaient fortement réduites ou absentes chez les doubles knockouts. Fait important, des contrôles détaillés de la composition corporelle, de la densité osseuse, de la fonction cardiaque, des marqueurs immunitaires et de la chimie sanguine n’ont révélé aucun problème non neurologique majeur causé par le blocage de cette étape précoce de la dégradation de la lysine.
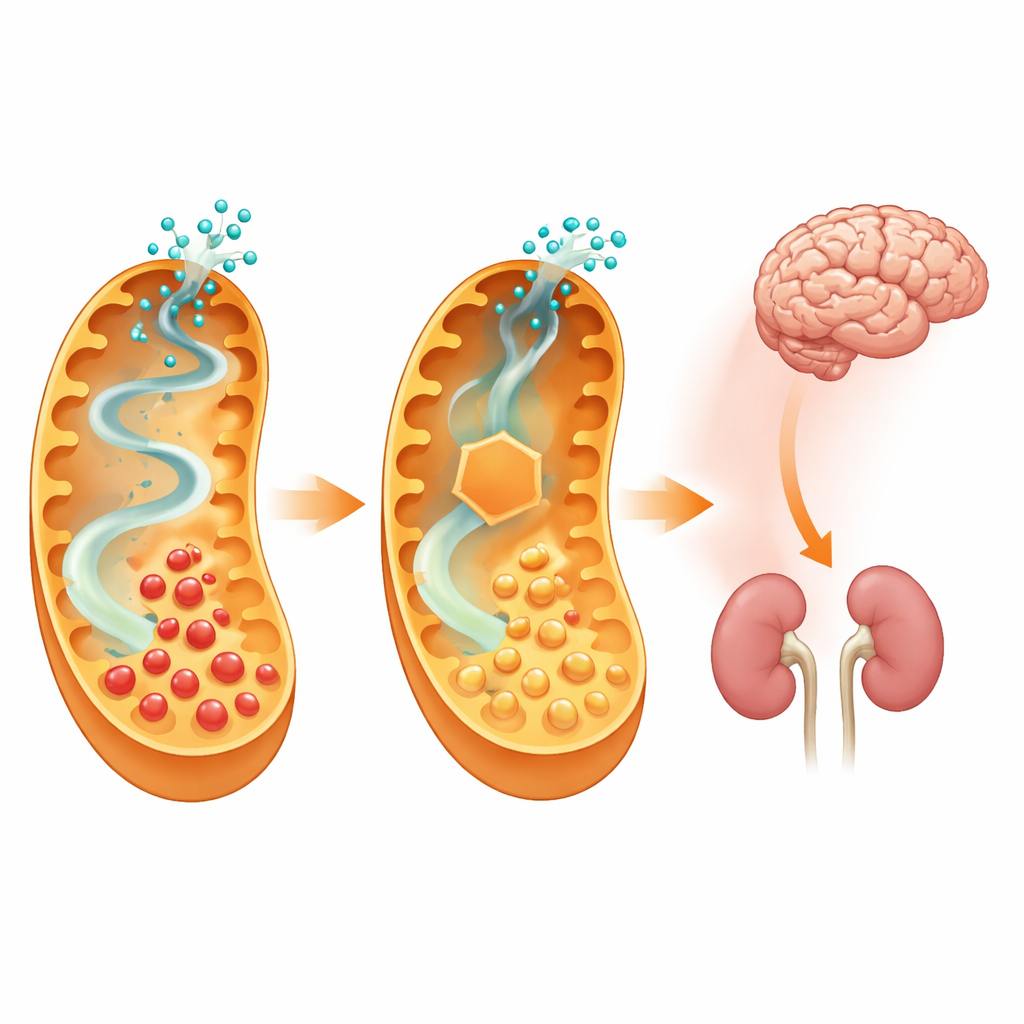
Ce que cela signifie pour les traitements futurs
En interrompant la première étape d’une voie spécifique de la lysine, les chercheurs ont pu réduire l’exposition du cerveau aux métabolites toxiques et protéger en grande partie les souris d’une poussée sévère de la maladie déclenchée par l’alimentation, sans causer de nouveaux problèmes de santé évidents. Cela suggère que des médicaments conçus pour inhiber partiellement la synthase de l’aminoadipate‑semialdéhyde pourraient offrir un nouvel outil puissant, en complément du régime et d’autres traitements, pour les enfants atteints d’acidurie glutarique de type 1. Bien que le travail actuel repose sur l’ingénierie génétique chez la souris, et que des médicaments appropriés doivent encore être développés et testés pour leur sécurité chez l’homme, l’étude fournit une forte preuve de principe que le blocage ciblé en amont de la chimie métabolique peut modifier le cours d’une maladie cérébrale métabolique dévastatrice.
Citation: Saad, C., Jung-Klawitter, S., Dimitrov, B. et al. Aminoadipate-semialdehyde synthase, a potential target for substrate reduction therapy in glutaric aciduria type 1. Sci Rep 16, 10995 (2026). https://doi.org/10.1038/s41598-026-44377-9
Mots-clés: acidurie glutarique de type 1, métabolisme de la lysine, thérapie de réduction du substrat, inhibition de AASS, troubles métaboliques héréditaires