Clear Sky Science · de
Aminoadipat-semialdehyd-Synthase, ein potenzielles Ziel für Substratreduktions-Therapie bei Glutarazidurie Typ 1
Warum diese seltene Kinderkrankheit wichtig ist
Die Glutarazidurie Typ 1 ist eine seltene erbliche Erkrankung, die vor allem Säuglinge und Kleinkinder betrifft. Sie veranschaulicht jedoch eine allgemeinere Idee: Wie das Verstellen eines einzelnen Schritts in der Körperchemie das Gehirn vor lebenslangen Schäden schützen kann. Trotz moderner Neugeborenen-Screenings und strenger Diäten entwickeln viele betroffene Kinder weiterhin Bewegungsstörungen, strukturelle Veränderungen im Gehirn und Nierenprobleme. Diese Studie untersucht einen neuen Ansatz, die Bildung schädlicher Substanzen bereits an der Quelle zu unterbrechen — eine Strategie, die Substratreduktions-Therapie genannt wird — und prüft, ob sie zu sichereren und wirksameren Behandlungen führen könnte.
Ein Verkehrschaos in der Körperchemie
Bei der Glutarazidurie Typ 1 kann der Körper bestimmte Proteinbausteine, insbesondere die Aminosäure Lysin, nicht richtig abbauen. Ein fehlendes oder fehlerhaftes Enzym, bekannt als Glutaryl‑CoA‑Dehydrogenase, verursacht einen metabolischen Stau, der zur Anreicherung von Glutarsäure und verwandten Verbindungen führt. Diese Substanzen sind insbesondere für bestimmte Hirnregionen, die Bewegungen steuern, besonders toxisch — vor allem in den ersten Lebensjahren. Selbst wenn Kinder kurz nach der Geburt erkannt und auf spezielle lysinarme Diäten gesetzt werden, entwickelt etwa ein Drittel schwere neurologische Symptome, und viele zeigen im Verlauf zunehmende Veränderungen der weißen Hirnsubstanz und Nierenprobleme.
Ein neuer Ort für eine Sperre
Die bisherige Behandlung zielt darauf ab, die Menge an aufgenommener Lysin zu begrenzen und dem Körper beim Abbau bzw. der Ausscheidung toxischer Nebenprodukte zu helfen, verändert aber nicht direkt den fehlerhaften Stoffwechselweg selbst. Die Forschenden stellten deshalb eine zentrale Frage: Anstatt mit dem angesammelten Abfall umzugehen, könnte man den Zufluss in den fehlerhaften Weg verlangsamen? Sie richteten ihren Blick auf ein anderes Enzym, die Aminoadipat‑semialdehyd‑Synthase (AASS), die die ersten beiden Schritte eines der Hauptabbauwege für Lysin katalysiert. Menschen, denen dieses Enzym natürlicherweise fehlt, haben typischerweise hohe Lysinwerte, aber wenige oder keine gesundheitlichen Probleme, was nahelegt, dass dessen Blockade relativ sicher sein könnte.
Das Konzept an gezüchteten Mäusen testen
Um diese Strategie zu prüfen, verwendete das Team Mäuse, denen die Glutaryl‑CoA‑Dehydrogenase fehlte — ein etabliertes Tiermodell der menschlichen Erkrankung. Diese Tiere reichern große Mengen toxischer Säuren in ihren Geweben an und entwickeln bei einer lysinreichen Diät Krampfanfälle, Bewegungsstörungen, Hirnschäden und oft tödliche Verläufe. Die Forschenden erzeugten anschließend Mäuse mit doppeltem Knockout, denen sowohl das Krankheitsenzym als auch die Aminoadipat‑semialdehyd‑Synthase fehlten. Unter normalen Fütterungsbedingungen erschienen diese Doppel‑Knockout‑Mäuse in einem breiten Testumfang gesund und verhielten sich wie normale Tiere, während sie nur milde biochemische Auffälligkeiten zeigten.
Weniger toxische Anreicherung und bessere Gehirngesundheit
Der eigentliche Test folgte, als beide Typen der Krankheitsmodelle einer lysinreichen Diät ausgesetzt wurden, um den anfälligen Stoffwechselweg zu stressen. Die nur das Krankheitsenzym fehlenden Mäuse häuften sehr hohe Konzentrationen toxischer Säuren im Gehirn, in Leber, Niere, Blut und Urin an, und viele entwickelten schwere neurologische Symptome, Krampfanfälle und Gewichtsverlust. Im Gegensatz dazu wiesen die Doppel‑Knockout‑Tiere in all diesen Geweben deutlich niedrigere Glutarsäurespiegel, weit weniger Krankheitszeichen und nahezu normales Verhalten auf. Die mikroskopische Untersuchung ihrer Gehirne zeigte, dass die kleinen Vakuolen und strukturellen Veränderungen, die bei den Einzel‑Knockout‑Mäusen sichtbar waren, bei den Doppel‑Knockouts stark reduziert oder nicht vorhanden waren. Wichtig ist, dass detaillierte Kontrollen von Körperzusammensetzung, Knochendichte, Herzfunktion, Immunparametern und Blutchemie keine größeren nicht‑neurologischen Probleme fanden, die durch die Blockade dieses frühen Schritts im Lysinabbau verursacht würden.
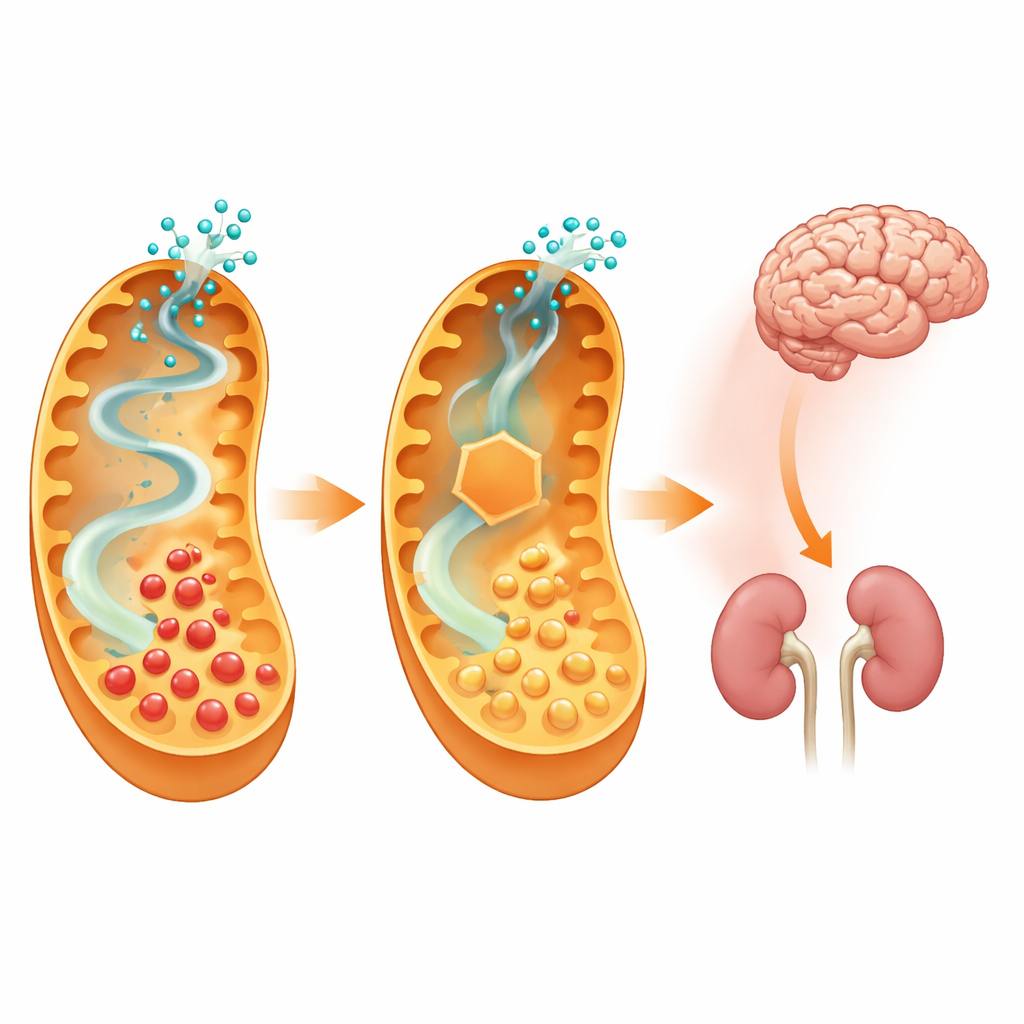
Was das für zukünftige Behandlungen bedeutet
Indem die Forschenden den ersten Schritt eines spezifischen Lysinabbauwegs ausschalteten, konnten sie die Exposition des Gehirns gegenüber toxischen Metaboliten senken und Mäuse weitgehend vor einem schweren, diätbedingt ausgelösten Krankheitsschub schützen, ohne offensichtliche neue Gesundheitsprobleme zu verursachen. Das deutet darauf hin, dass Arzneimittel, die eine teilweise Hemmung der Aminoadipat‑semialdehyd‑Synthase anstreben, ein wirkungsvolles neues Werkzeug neben Diät und anderen Behandlungen für Kinder mit Glutarazidurie Typ 1 sein könnten. Obwohl die aktuelle Arbeit auf genetischer Manipulation bei Mäusen beruht und geeignete Medikamente noch entwickelt sowie auf ihre Sicherheit beim Menschen geprüft werden müssen, liefert die Studie einen starken Proof‑of‑Principle, dass eine gezielte Blockade des vorgelagerten Stoffwechsels den Verlauf einer verheerenden metabolischen Hirnerkrankung verändern kann.
Zitation: Saad, C., Jung-Klawitter, S., Dimitrov, B. et al. Aminoadipate-semialdehyde synthase, a potential target for substrate reduction therapy in glutaric aciduria type 1. Sci Rep 16, 10995 (2026). https://doi.org/10.1038/s41598-026-44377-9
Schlüsselwörter: Glutarazidurie Typ 1, Lysin-Stoffwechsel, Substratreduktions-Therapie, AASS-Hemmung, vererbte Stoffwechselerkrankungen