Clear Sky Science · it
Sintasi dell’aminoadipato-semialdeide, un potenziale bersaglio per la terapia di riduzione del substrato nella glutaric aciduria di tipo 1
Perché questa rara malattia infantile conta
La glutaric aciduria di tipo 1 è una rara malattia ereditaria che colpisce principalmente neonati e bambini piccoli, ma illustra un’idea molto più ampia: come modificare un singolo passaggio nella chimica del corpo possa proteggere il cervello da danni permanenti. Nonostante gli screening neonatali moderni e diete attente, molti bambini con questa condizione sviluppano comunque problemi del movimento, alterazioni cerebrali e problemi renali. Questo studio esplora un nuovo modo per interrompere la produzione di sostanze nocive alla fonte, usando una strategia chiamata terapia di riduzione del substrato, e verifica se potrebbe portare a trattamenti più sicuri ed efficaci.
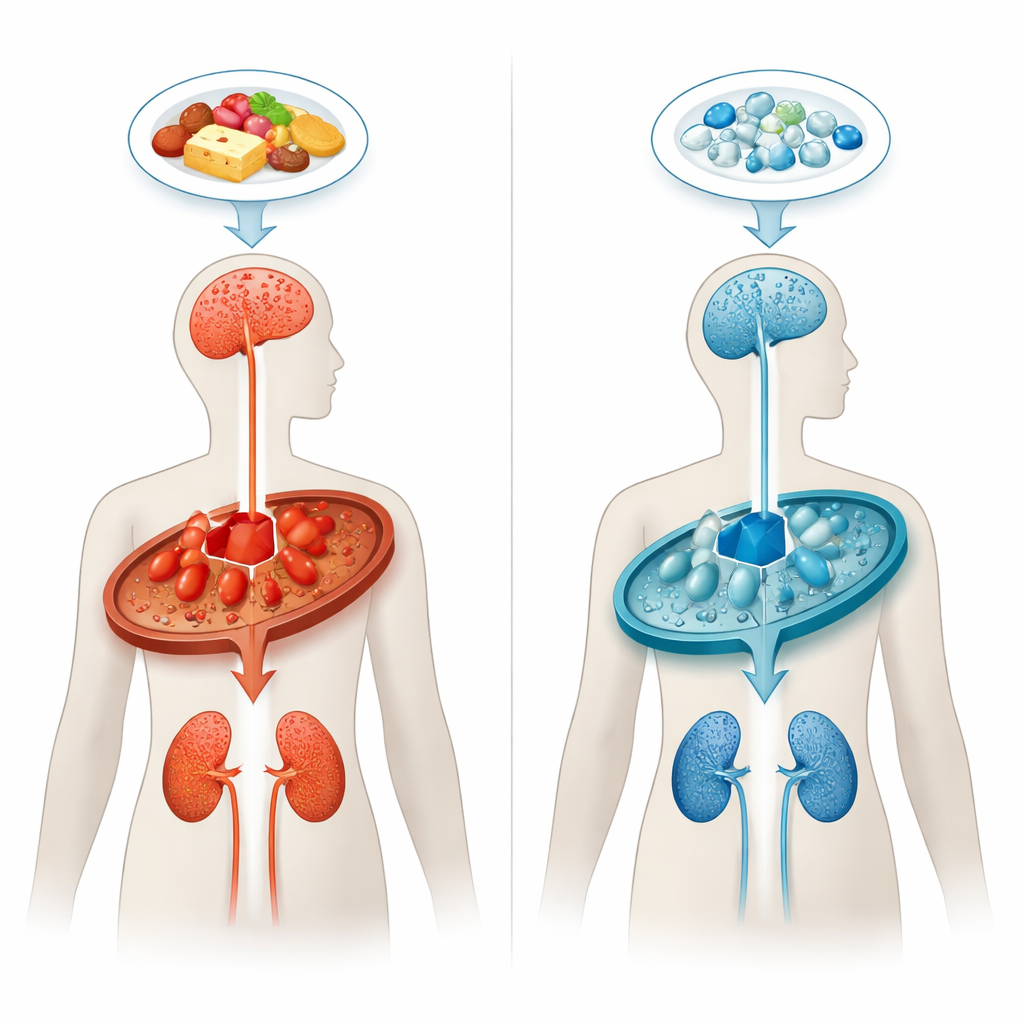
Un ingorgo nel traffico chimico dell’organismo
Nella glutaric aciduria di tipo 1, l’organismo non riesce a degradare correttamente alcuni mattoni delle proteine, in particolare l’amminoacido lisina. Una carenza o un malfunzionamento dell’enzima noto come glutaril-CoA deidrogenasi provoca un ingorgo metabolico, con accumulo di acido glutarico e composti correlati. Queste sostanze sono particolarmente tossiche per regioni cerebrali specifiche che controllano il movimento, soprattutto nelle fasi precoci della vita. Anche quando i bambini vengono identificati poco dopo la nascita e messi a diete speciali a basso contenuto di lisina, circa uno su tre sviluppa ancora sintomi neurologici gravi, e molti mostrano progressivi problemi della sostanza bianca e dei reni con l’età.
Un nuovo punto dove porre un blocco
Il trattamento attuale si concentra sul limitare la quantità di lisina che entra nel sistema e sull’aiutare l’organismo a rimuovere i prodotti tossici, ma non modifica direttamente il funzionamento del percorso difettoso. I ricercatori si sono posti una domanda chiave: invece di cercare di gestire i rifiuti accumulati, potremmo rallentare l’ingresso nel percorso difettoso stesso? Hanno focalizzato l’attenzione su un altro enzima chiamato sintasi dell’aminoadipato-semialdeide, che esegue i primi due passaggi in una delle principali vie di degradazione della lisina. Le persone che in modo naturale sono carenti di questo enzima presentano tipicamente livelli elevati di lisina ma pochi o nessun problema di salute, suggerendo che bloccarlo potrebbe essere relativamente sicuro.
Testare l’idea in topi ingegnerizzati
Per sondare questa strategia, il team ha usato topi allevati per essere privi della glutaril-CoA deidrogenasi, un modello ampiamente consolidato della malattia umana. Questi animali accumulano grandi quantità di acidi tossici nei tessuti e, quando sottoposti a una dieta ricca di lisina, sviluppano convulsioni, problemi del movimento, danni cerebrali e spesso muoiono. I ricercatori hanno quindi creato un topo doppio knock-out privo sia dell’enzima della malattia sia della sintasi dell’aminoadipato-semialdeide. In condizioni alimentari standard, questi topi doppio knock-out apparivano e si comportavano come animali sani in una vasta batteria di test, mostrando al contempo solo lievi anomalie biochimiche.
Meno accumulo tossico e migliore salute cerebrale
La prova decisiva è arrivata quando entrambi i tipi di topi modello della malattia sono stati esposti a una dieta ad alto contenuto di lisina progettata per sollecitare il percorso vulnerabile. I topi mancanti solo dell’enzima della malattia accumulavano livelli molto elevati di acidi tossici nel cervello, nel fegato, nei reni, nel sangue e nelle urine, e molti sviluppavano gravi sintomi neurologici, convulsioni e perdita di peso. Al contrario, gli animali doppio knock-out presentavano livelli di acido glutarico drasticamente più bassi in tutti questi tessuti, segni di malattia molto ridotti e un comportamento quasi normale. L’esame microscopico dei loro cervelli ha mostrato che le piccole vacuolizzazioni e le modificazioni strutturali osservate nei topi knock-out singoli erano notevolmente ridotte o assenti nei doppio knock-out. È importante sottolineare che controlli dettagliati della composizione corporea, della densità ossea, della funzione cardiaca, dei marcatori immunitari e della chimica ematica non hanno evidenziato problemi non neurologici rilevanti causati dal bloccare questo passaggio precoce nella degradazione della lisina.
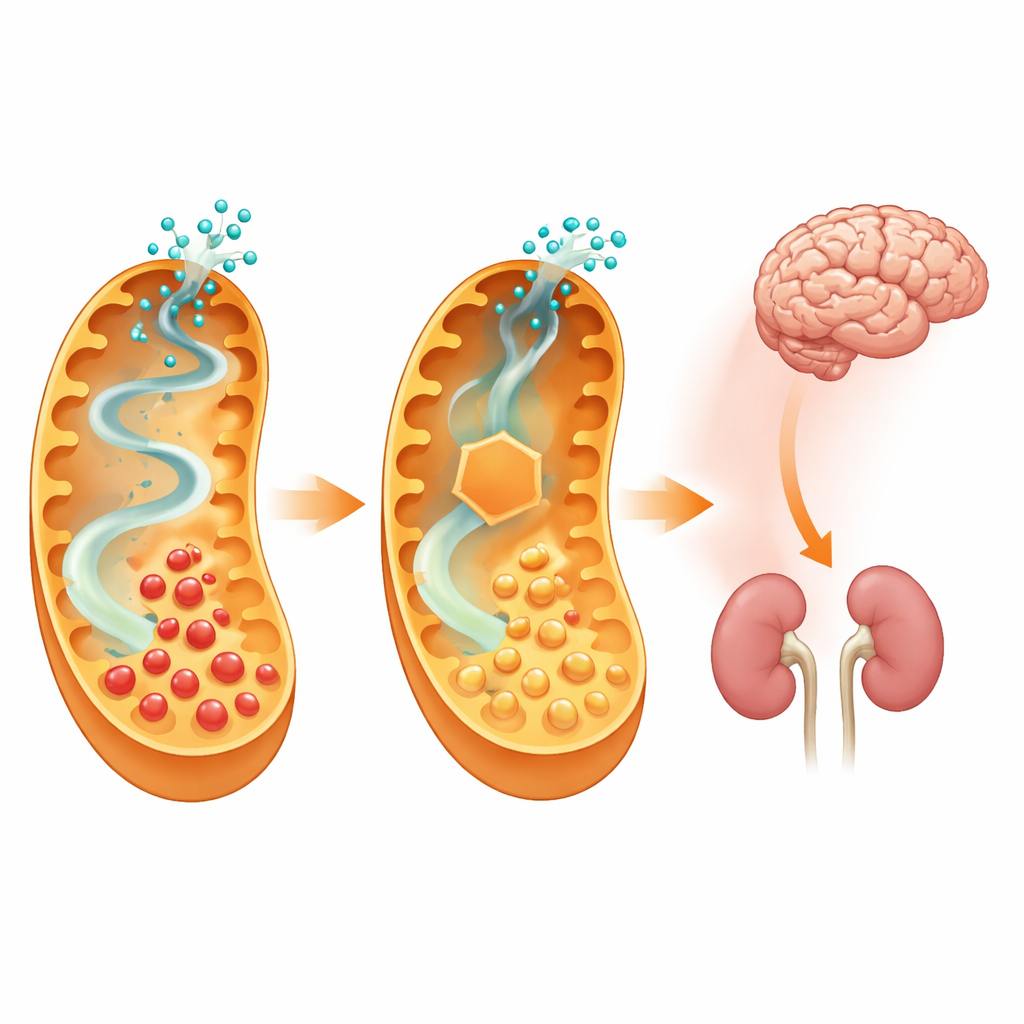
Cosa significa per i trattamenti futuri
Spegnendo il primo passaggio di una specifica via della lisina, i ricercatori sono riusciti a ridurre l’esposizione del cervello a metaboliti tossici e a proteggere in larga misura i topi da una grave riacutizzazione della malattia scatenata dalla dieta, senza causare evidenti nuovi problemi di salute. Ciò suggerisce che farmaci progettati per inibire parzialmente la sintasi dell’aminoadipato-semialdeide potrebbero offrire uno strumento potente da affiancare a dieta e altri trattamenti per i bambini con glutaric aciduria di tipo 1. Sebbene il lavoro attuale si basi sull’ingegneria genetica nei topi, e sia ancora necessario sviluppare farmaci adeguati e testarli per la sicurezza negli esseri umani, lo studio fornisce una solida prova di principio che un blocco mirato della chimica a monte può rimodellare l’evoluzione di una devastante malattia metabolica cerebrale.
Citazione: Saad, C., Jung-Klawitter, S., Dimitrov, B. et al. Aminoadipate-semialdehyde synthase, a potential target for substrate reduction therapy in glutaric aciduria type 1. Sci Rep 16, 10995 (2026). https://doi.org/10.1038/s41598-026-44377-9
Parole chiave: glutaric aciduria di tipo 1, metabolismo della lisina, terapia di riduzione del substrato, inibizione di AASS, malattie metaboliche ereditarie