Clear Sky Science · pl
Rurociąg metod łączenia danych wielomodalnych opartych na uczeniu maszynowym do analizy ryzyka prognostycznego u pacjentów z przerzutowym rakiem jelita grubego leczonych bewacyzumabem
Dlaczego to badanie ma znaczenie
Dla osób żyjących z zaawansowanym rakiem jelita grubego jednym z najważniejszych pytań jest, czy lek o dużej sile działania, ale wysokim koszcie, rzeczywiście im pomoże. Badanie to analizuje, jak wzorce w DNA guza pacjenta, w połączeniu z danymi klinicznymi, można wykorzystać za pomocą nowoczesnego uczenia maszynowego do przewidywania, kto prawdopodobnie skorzysta z powszechnie stosowanego leczenia celowanego — bewacyzumabu — a kto nie. W przyszłości takie narzędzia mogłyby uchronić niektórych pacjentów przed skutkami ubocznymi i nieskuteczną terapią, a innych skierować ku najbardziej obiecującym opcjom.
Bliższe spojrzenie na leczenie raka jelita
Przerzutowy rak jelita grubego — rak, który rozprzestrzenił się do innych narządów — jest główną przyczyną zgonów z powodu nowotworów na świecie. Wielu pacjentów, których guzy mają określone zmiany genowe (mutacje w genach RAS), otrzymuje standardową chemioterapię w połączeniu z bewacyzumabem, lekiem blokującym wzrost naczyń krwionośnych, by „zagłodzić” guz. Choć to skojarzenie przeciętnie poprawia przeżycie, tylko część pacjentów odnosi znaczącą korzyść. Inni przechodzą miesiące leczenia, doświadczają działań niepożądanych i ponoszą koszty finansowe przy niewielim zysku. Obecnie lekarze nie dysponują niezawodnym testem, który pozwoliłby wcześniej stwierdzić, kto nie odpowie na bewacyzumab, co tworzy pilną potrzebę lepszych narzędzi decyzyjnych.
Łączenie wielu typów danych
Naukowcy zbudowali wieloetapowy pipeline analityczny, który wykorzystuje uczenie maszynowe do integracji kilku rodzajów informacji o każdym pacjencie. Skorzystali z dobrze opisanej europejskiej kohorty o nazwie ANGIOPREDICT, obejmującej 117 osób z przerzutowym rakiem jelita grubego leczonych bewacyzumabem w połączeniu z chemioterapią. Dla każdego pacjenta posiadali: regiony genomu wykazujące zyski lub utraty kopii (zmiany liczby kopii), niewielki zestaw istotnych mutacji genowych oraz standardowe dane kliniczne, takie jak wiek, stadium guza i lokalizacja nowotworu. Specjalistyczne narzędzie o nazwie PhenMap posłużyło następnie do odkrycia ukrytych wzorców — tzw. meta-zmiennych — które podsumowują, jak te zmiany genetyczne i cechy kliniczne współwystępują u poszczególnych pacjentów.
Wyszukiwanie sygnatury DNA powiązanej z wynikiem
Wśród dziesięciu wzorców zidentyfikowanych przez PhenMap dwa były silnie związane z długością czasu, przez jaki pacjenci żyli bez pogorszenia choroby — miarą zwaną przeżyciem wolnym od progresji (PFS). Zespół skupił się następnie na tym, które konkretne zmiany w DNA odpowiadały za te dwa kluczowe wzorce. Dzięki dodatkowym krokom statystycznym i uczenia maszynowego zawęzili setki regionów genomowych i mutacji do zaledwie trzech cech: utrat w dwóch regionach chromosomalnych (15q21.1 i 1p36.31) oraz mutacji w genie BRAF. Te trzy cechy razem utworzyły zwięzłą sygnaturę genetyczną ściśle związaną z gorszymi wynikami u pacjentów leczonych bewacyzumabem.
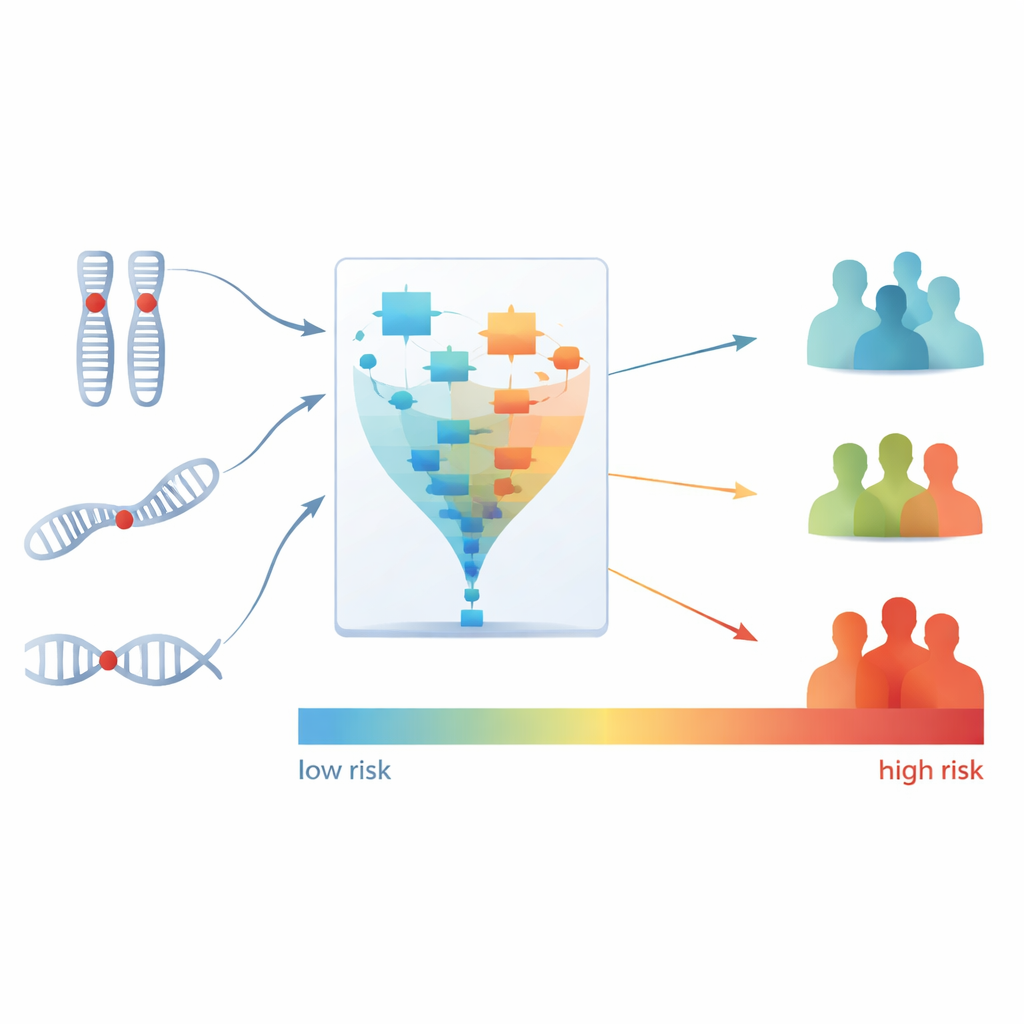
Przekształcenie sygnatury w grupy ryzyka
Następnie naukowcy zamienili tę trzyczęściową sygnaturę na pojedynczy wynik ryzyka dla każdego pacjenta, odzwierciedlający szacowane ryzyko zgonu podczas terapii opartej na bewacyzumabie. Podzielili potem pacjentów na trzy grupy — niskiego, średniego i wysokiego ryzyka — na podstawie tych wyników. Różnice były uderzające: każdy pacjent z grupy wysokiego ryzyka nie odpowiedział na bewacyzumab, podczas gdy większość pacjentów z grupy niskiego ryzyka wykazała odpowiedź. Grupa wysokiego ryzyka miała także znacznie większe prawdopodobieństwo wczesnej progresji choroby w porównaniu z grupą niskiego ryzyka. Co ważne, ten wynik ryzyka dostarczał informacji prognostycznych wykraczających poza to, co lekarze mogli już wywnioskować z standardowych czynników klinicznych lub samego wcześniejszego podtypowania genomowego.
Co to może oznaczać dla pacjentów
Choć wyniki te wymagają jeszcze walidacji w większych i niezależnych kohortach pacjentów, wskazują na przyszłość, w której złożone dane o guzie i dane kliniczne można zintegrować w pojedynczym, praktycznym wyniku ryzyka. Jeśli zostanie to potwierdzone, prosty test wykrywający obecność dwóch utrat chromosomalnych i mutacji BRAF mógłby pomóc zidentyfikować pacjentów z przerzutowym rakiem jelita grubego, którzy nie odniosą korzyści ze skojarzonej terapii z bewacyzumabem. Tacy pacjenci mogliby zostać wcześniej skierowani do alternatywnych strategii lub badań klinicznych, podczas gdy inni kontynuowaliby leczenie, z którego mają większe szanse skorzystać. Szerzej rzecz biorąc, pokazany tu pipeline uczenia maszynowego można by dostosować do innych nowotworów i terapii, przybliżając cel prawdziwie spersonalizowanej opieki onkologicznej.
Cytowanie: Thomas, V., Nyamundanda, G., Lärkeryd, A. et al. A pipeline of machine learning-driven multi-modal data fusion methods for prognostic risk analysis in bevacizumab-treated metastatic colorectal cancer. Sci Rep 16, 8843 (2026). https://doi.org/10.1038/s41598-026-39189-w
Słowa kluczowe: przerzutowy rak jelita grubego, oporność na bewacyzumab, uczenie maszynowe, biomarkery genomowe, onkolgia precyzyjna