Clear Sky Science · fr
Une chaîne de méthodes de fusion de données multimodales pilotée par apprentissage automatique pour l’analyse pronostique du risque chez des patients atteints de cancer colorectal métastatique traités par bevacizumab
Pourquoi cette recherche est importante
Pour les personnes vivant avec un cancer du côlon avancé, l’une des questions majeures est de savoir si un médicament puissant mais coûteux leur apportera réellement un bénéfice. Cette étude examine comment les motifs présents dans l’ADN tumoral d’un patient, combinés aux informations cliniques, peuvent être utilisés avec les méthodes modernes d’apprentissage automatique pour prédire qui est susceptible de bénéficier d’un traitement ciblé courant, le bevacizumab, et qui ne le sera pas. À l’avenir, de tels outils pourraient épargner à certains patients des effets secondaires et des traitements inefficaces, tout en orientant d’autres vers les options les plus prometteuses.
Un regard plus précis sur le traitement du cancer colorectal
Le cancer colorectal métastatique — un cancer du côlon qui s’est propagé à d’autres organes — est une cause majeure de mortalité par cancer dans le monde. De nombreux patients dont les tumeurs portent des altérations génétiques spécifiques (mutations RAS) reçoivent une chimiothérapie standard associée au bevacizumab, un médicament qui bloque la croissance des vaisseaux sanguins pour priver les tumeurs de leur apport. Bien que cette combinaison améliore la survie en moyenne, seule une fraction des patients en tire un bénéfice significatif. D’autres subissent des mois de traitement, des effets secondaires et des coûts financiers avec peu de gain. À l’heure actuelle, les médecins ne disposent d’aucun test fiable permettant de savoir à l’avance qui ne répondra pas au bevacizumab, ce qui crée un besoin pressant d’outils décisionnels meilleurs.
Combiner plusieurs types de données
Les chercheurs ont construit une chaîne d’analyse en plusieurs étapes qui utilise l’apprentissage automatique pour fusionner plusieurs types d’informations pour chaque patient. Ils se sont appuyés sur une cohorte européenne bien caractérisée appelée ANGIOPREDICT, qui comprend 117 personnes atteintes de cancer colorectal métastatique traitées par bevacizumab et chimiothérapie. Pour chaque patient, ils disposaient : de régions du génome présentant des gains ou des pertes (altérations du nombre de copies), d’un petit ensemble de mutations géniques importantes, et d’éléments cliniques standard tels que l’âge, le stade tumoral et la localisation de la tumeur. Un outil spécialisé appelé PhenMap a ensuite été utilisé pour découvrir des motifs cachés — appelés méta-variables — qui résument la manière dont ces modifications génétiques et ces caractéristiques cliniques varient ensemble entre les patients.
Identifier la signature ADN liée au pronostic
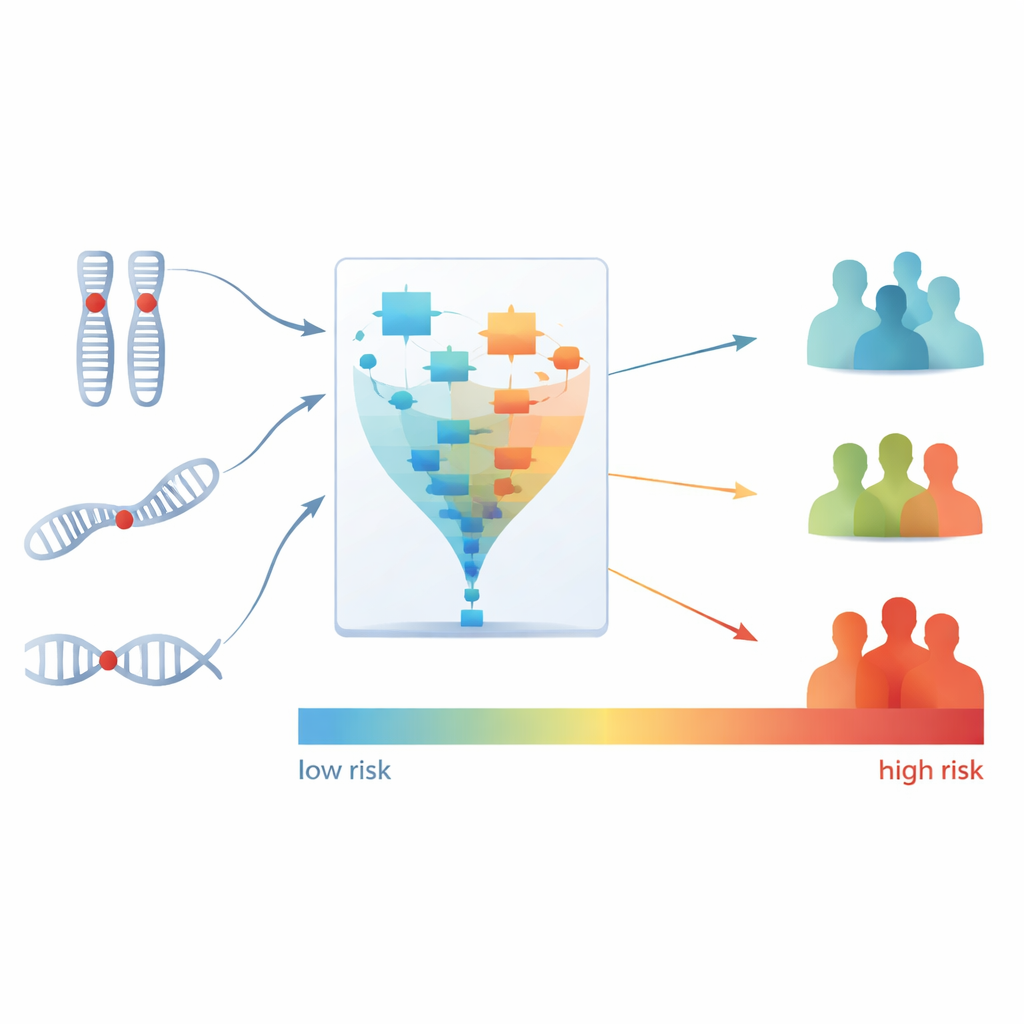
Parmi les dix motifs identifiés par PhenMap, deux étaient fortement liés à la durée pendant laquelle les patients ont vécu sans progression de la maladie, une mesure appelée survie sans progression. L’équipe s’est ensuite concentrée sur les modifications d’ADN spécifiques qui sous-tendaient ces deux motifs clés. À l’aide d’étapes supplémentaires statistiques et d’apprentissage automatique, ils ont réduit des centaines de régions génomiques et mutations à seulement trois caractéristiques : des pertes dans deux régions chromosomiques (15q21.1 et 1p36.31) et une mutation dans un gène appelé BRAF. Ces trois caractéristiques formaient ensemble une signature génétique compacte étroitement liée à de moins bons résultats chez les patients ayant reçu du bevacizumab.
Transformer une signature en groupes de risque
Ensuite, les scientifiques ont converti cette signature en trois éléments en un score de risque unique pour chaque patient, reflétant leur risque estimé de décès sous traitement à base de bevacizumab. Ils ont ensuite divisé les patients en trois groupes — faible, moyen et élevé risque — en fonction de leurs scores. Les différences étaient frappantes : tous les patients du groupe à haut risque n’ont pas répondu au bevacizumab, tandis que la majeure partie des patients du groupe à faible risque montraient une réponse. Le groupe à haut risque présentait également une probabilité beaucoup plus élevée de progression précoce de la maladie comparé au groupe à faible risque. Fait important, ce score de risque apportait une information pronostique supplémentaire à celle que les médecins pouvaient déjà déduire des facteurs cliniques standard ou du seul sous-typage génomique antérieur.
Ce que cela pourrait signifier pour les patients
Bien que ce travail doive encore être validé dans des cohortes de patients plus larges et indépendantes, il ouvre la voie à un avenir dans lequel des données tumorales et cliniques complexes peuvent être intégrées dans un score de risque unique et exploitable. Si cela est confirmé, un test simple indiquant la présence des deux pertes chromosomiques et de la mutation BRAF pourrait aider à identifier les patients atteints de cancer colorectal métastatique peu susceptibles de bénéficier de la thérapie combinée au bevacizumab. Ces patients pourraient alors être orientés plus tôt vers des stratégies alternatives ou des essais cliniques, tandis que d’autres continueraient à recevoir un médicament dont ils ont davantage de chances de tirer profit. Plus largement, la chaîne d’apprentissage automatique démontrée ici pourrait être adaptée à d’autres cancers et traitements, faisant progresser l’objectif d’un soin véritablement personnalisé du cancer.
Citation: Thomas, V., Nyamundanda, G., Lärkeryd, A. et al. A pipeline of machine learning-driven multi-modal data fusion methods for prognostic risk analysis in bevacizumab-treated metastatic colorectal cancer. Sci Rep 16, 8843 (2026). https://doi.org/10.1038/s41598-026-39189-w
Mots-clés: cancer colorectal métastatique, résistance au bevacizumab, apprentissage automatique, biomarqueurs génomiques, oncologie de précision