Clear Sky Science · de
Eine Pipeline maschinell lerngetriebener multimodaler Datenfusionsmethoden zur prognostischen Risikoanalyse bei mit Bevacizumab behandelten metastasierten Kolorektalkarzinomen
Warum diese Forschung wichtig ist
Für Menschen mit fortgeschrittenem Darmkrebs zählt zu den drängendsten Fragen, ob ein wirksames, aber teures Medikament ihnen tatsächlich nützt. Diese Studie untersucht, wie Muster in der Tumor-DNA eines Patienten zusammen mit klinischen Informationen mithilfe moderner Verfahren des maschinellen Lernens genutzt werden können, um vorherzusagen, wer wahrscheinlich von einer verbreiteten zielgerichteten Therapie, Bevacizumab, profitiert und wer nicht. Solche Werkzeuge könnten künftig einige Patienten vor Nebenwirkungen und unwirksamer Therapie bewahren und andere zu den vielversprechendsten Optionen führen.
Ein genauerer Blick auf die Behandlung von Darmkrebs
Metastasiertes Kolorektalkarzinom — Darmkrebs, der in andere Organe gestreut hat — ist weltweit eine bedeutende Todesursache durch Krebs. Viele Patienten, deren Tumoren bestimmte Genveränderungen (RAS-Mutationen) tragen, erhalten eine Standardchemotherapie in Kombination mit Bevacizumab, einem Medikament, das das Wachstum von Blutgefäßen blockiert, um Tumore zu „verhungern“. Obwohl diese Kombination im Mittel das Überleben verbessert, profitiert nur ein Teil der Patienten wirklich merklich. Andere durchlaufen Monate der Behandlung mit Nebenwirkungen und hohen Kosten, ohne nennenswerten Nutzen. Derzeit gibt es für Ärzte keinen verlässlichen Test, der im Voraus aussagt, wer nicht auf Bevacizumab anspricht, weshalb bessere Entscheidungswerkzeuge dringend benötigt werden.
Verschiedene Datentypen zusammenführen
Die Forschenden entwickelten eine mehrstufige Analyse-Pipeline, die maschinelles Lernen einsetzt, um mehrere Arten von Informationen pro Patient zu integrieren. Sie nutzten einen gut charakterisierten europäischen Kohortenbestand namens ANGIOPREDICT, der 117 Menschen mit metastasiertem Kolorektalkarzinom umfasst, die mit Bevacizumab plus Chemotherapie behandelt wurden. Für jeden Patienten lagen vor: Regionen des Genoms, die Gewinne oder Verluste zeigten (Copy-Number-Alterationen), eine kleine Auswahl wichtiger Genmutationen sowie standardmäßige klinische Angaben wie Alter, Tumorstadium und Tumorlage. Ein spezialisiertes Werkzeug namens PhenMap wurde dann verwendet, um verborgene Muster — sogenannte Meta-Variablen — zu entdecken, die zusammenfassen, wie diese genetischen Veränderungen und klinischen Merkmale über die Patienten hinweg gemeinsam variieren.
Die DNA-Signatur, die mit dem Ergebnis verknüpft ist, finden
Unter den zehn von PhenMap identifizierten Mustern standen zwei in starkem Zusammenhang mit der Dauer, die Patienten lebten, ohne dass ihre Erkrankung sich verschlechterte — einem Maß namens progressionsfreies Überleben. Das Team konzentrierte sich anschließend darauf, welche spezifischen DNA-Veränderungen diese beiden Schlüsselmuster antrieben. Mithilfe zusätzlicher statistischer und maschineller Lernverfahren reduzierten sie Hunderte genomischer Regionen und Mutationen auf nur drei Merkmale: Verluste in zwei chromosomalen Regionen (15q21.1 und 1p36.31) sowie eine Mutation im Gen BRAF. Diese drei Merkmale bildeten zusammen eine kompakte genetische Signatur, die eng mit schlechteren Ergebnissen bei Patienten verbunden war, die Bevacizumab erhielten.
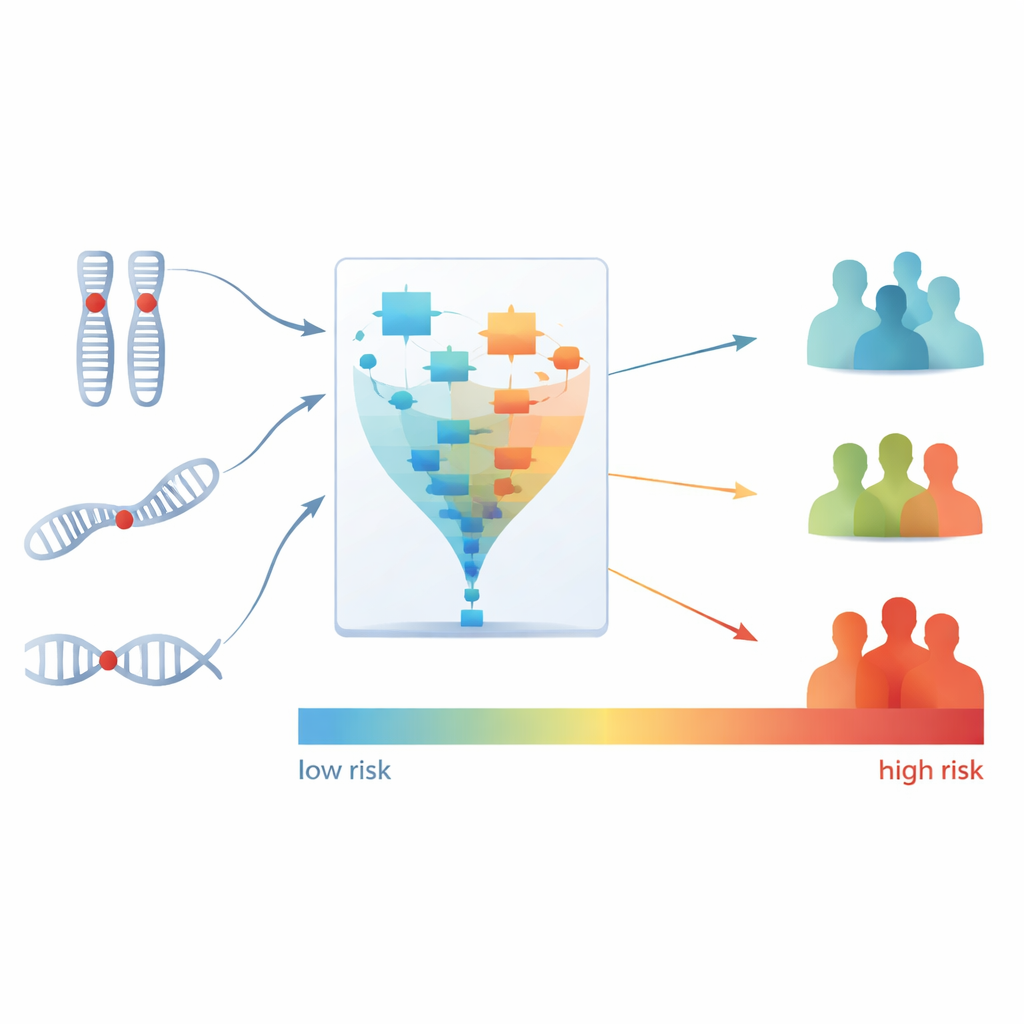
Eine Signatur in Risikogruppen umwandeln
Im nächsten Schritt wandten die Wissenschaftler diese dreiteilige Signatur an, um für jeden Patienten einen einzigen Risikoscore zu berechnen, der dessen geschätztes Sterberisiko unter einer Bevacizumab-basierten Therapie widerspiegelt. Sie unterteilten die Patienten dann anhand ihrer Scores in drei Gruppen — niedriges, mittleres und hohes Risiko. Die Unterschiede waren deutlich: Jeder Patient in der Hochrisikogruppe sprach nicht auf Bevacizumab an, während die meisten Patienten in der Niedrigrisikogruppe eine Ansprechrate zeigten. Die Hochrisikogruppe hatte zudem eine deutlich höhere Wahrscheinlichkeit für frühe Krankheitsprogression im Vergleich zur Niedrigrisikogruppe. Wichtig ist, dass dieser Risikoscore prognostische Informationen lieferte, die über das hinausgingen, was Ärzte bereits aus standardmäßigen klinischen Faktoren oder früheren genomischen Subtypen ableiten konnten.
Was das für Patienten bedeuten könnte
Obwohl diese Arbeit noch in größeren und unabhängigen Patientenkohorten validiert werden muss, deutet sie auf eine Zukunft hin, in der komplexe Tumor- und klinische Daten in einem einzigen, handhabbaren Risikoscore integriert werden können. Falls bestätigt, könnte ein einfacher Test, der das Vorliegen der beiden chromosomalen Verluste und der BRAF-Mutation erfasst, dabei helfen, Patienten mit metastasiertem Kolorektalkarzinom zu identifizieren, die wahrscheinlich nicht von einer Kombinationstherapie mit Bevacizumab profitieren. Diese Patienten könnten dann früher zu alternativen Strategien oder klinischen Studien weitergeleitet werden, während andere weiterhin ein Medikament erhalten, von dem sie eher Nutzen haben. Allgemeiner lässt sich die hier gezeigte Pipeline des maschinellen Lernens auf andere Krebsarten und Behandlungen anpassen und damit das Ziel einer tatsächlich personalisierten Krebsbehandlung voranbringen.
Zitation: Thomas, V., Nyamundanda, G., Lärkeryd, A. et al. A pipeline of machine learning-driven multi-modal data fusion methods for prognostic risk analysis in bevacizumab-treated metastatic colorectal cancer. Sci Rep 16, 8843 (2026). https://doi.org/10.1038/s41598-026-39189-w
Schlüsselwörter: metastasiertes Kolorektalkarzinom, Bevacizumab-Resistenz, maschinelles Lernen, genomische Biomarker, Präzisionsonkologie