Clear Sky Science · nl
Een pijplijn van machine learning-gedreven multimodale datafusie methoden voor prognostische risicoanalyse bij met bevacizumab behandelde metastatisch colorectaal carcinoom
Waarom dit onderzoek ertoe doet
Voor mensen met gevorderde darmkanker is een van de grootste vragen of een krachtig maar duur geneesmiddel hen daadwerkelijk zal helpen. Deze studie onderzoekt hoe patronen in het tumor-DNA van een patiënt, in combinatie met klinische gegevens, met moderne machine learning kunnen worden gebruikt om te voorspellen wie waarschijnlijk baat heeft bij een veelgebruikte gerichte behandeling, bevacizumab, en wie niet. In de toekomst zouden dergelijke hulpmiddelen sommige patiënten kunnen besparen van bijwerkingen en ineffectieve therapie, terwijl anderen naar de meest veelbelovende opties worden geleid.
Een nadere blik op de behandeling van darmkanker
Metastatisch colorectaal carcinoom — darmkanker die is uitgezaaid naar andere organen — is een belangrijke oorzaak van kankersterfte wereldwijd. Veel patiënten van wie de tumoren specifieke genetische veranderingen (RAS-mutaties) dragen, krijgen standaard chemotherapie gecombineerd met bevacizumab, een middel dat de bloedvatvorming blokkeert om tumoren uit te hongeren. Hoewel deze combinatie de overleving gemiddeld verbetert, ziet slechts een deel van de patiënten een wezenlijk voordeel. Anderen ondergaan maandenlange behandeling, bijwerkingen en financiële lasten met weinig winst. Op dit moment hebben artsen geen betrouwbare test om van tevoren te zeggen wie niet op bevacizumab zal reageren, wat de noodzaak voor betere beslissingsinstrumenten vergroot.
Verschillende datatypes samenbrengen
De onderzoekers bouwden een meerstappen-analysepijplijn die machine learning gebruikt om verschillende soorten informatie van elke patiënt te fusen. Ze putten uit een goed gekarakteriseerde Europese cohort genaamd ANGIOPREDICT, dat 117 mensen met metastatisch colorectaal carcinoom omvat die werden behandeld met bevacizumab plus chemotherapie. Voor elke patiënt hadden ze: regio’s in het genoom die ofwel gewonnen of verloren waren (copy number-alteraties), een kleine set belangrijke genmutaties, en standaard klinische gegevens zoals leeftijd, tumorniveau en tumorlocatie. Een gespecialiseerd hulpmiddel genaamd PhenMap werd vervolgens gebruikt om verborgen patronen te ontdekken — zogenaamde meta-variabelen — die samenvatten hoe deze genetische veranderingen en klinische kenmerken samen variëren tussen patiënten.
Het DNA-handtekening vinden die aan uitkomst gekoppeld is
Van de tien patronen die PhenMap identificeerde, waren er twee sterk verbonden met hoe lang patiënten leefden zonder dat hun ziekte verergerde, een maatstaf die progression-free survival wordt genoemd. Het team concentreerde zich daarna op welke specifieke DNA-veranderingen deze twee sleutelpatronen aandreven. Met aanvullende statistische en machine-learningstappen beperkten ze honderden genomische regio’s en mutaties tot slechts drie kenmerken: verliezen in twee chromosomale regio’s (15q21.1 en 1p36.31) en een mutatie in een gen dat BRAF heet. Deze drie kenmerken samen vormden een compacte genetische signatuur die sterk gekoppeld was aan slechtere uitkomsten bij patiënten die bevacizumab ontvingen.
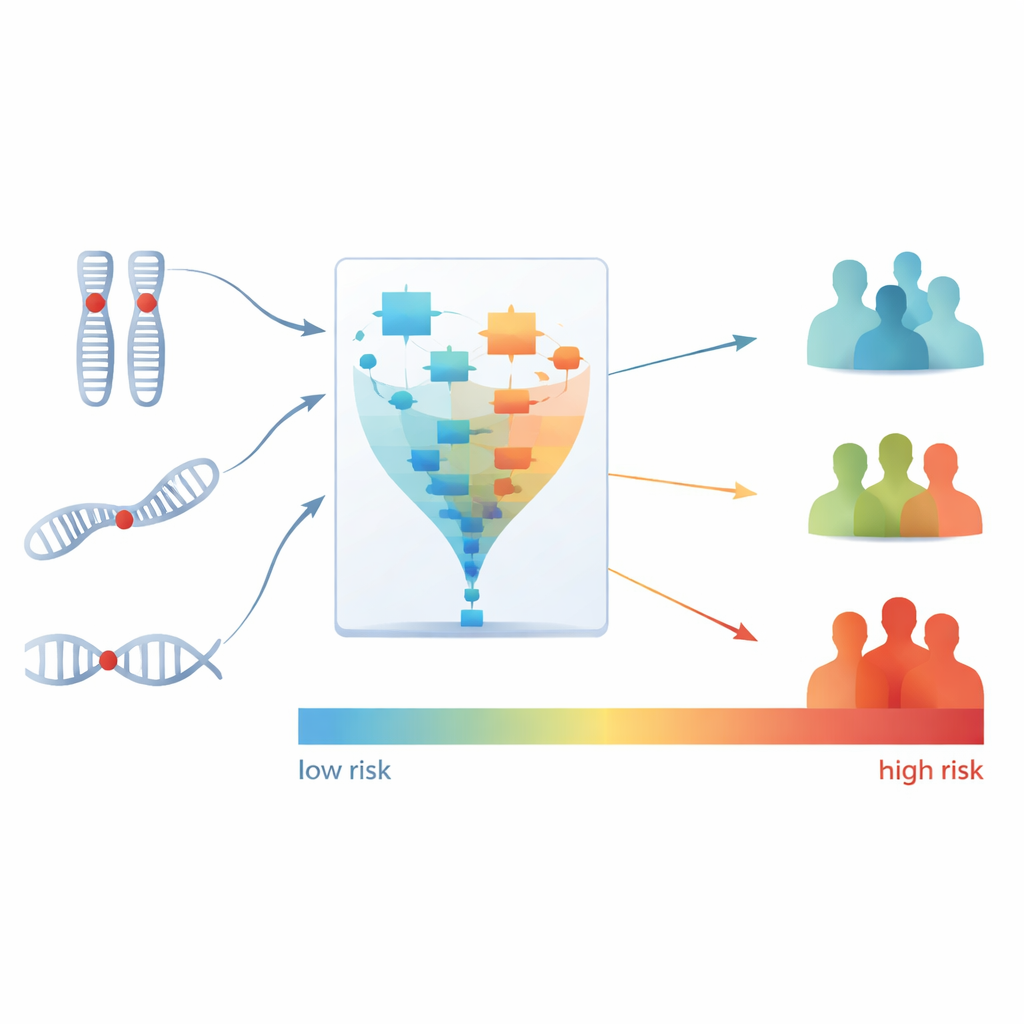
Een signatuur omzetten in risicogroepen
Vervolgens zetten de wetenschappers deze driedelige signatuur om in een enkele risicoscore voor elke patiënt, die hun geschatte risico op overlijden tijdens behandeling met bevacizumab-gebaseerde therapie weerspiegelt. Ze verdeelden patiënten daarna in drie groepen — laag, medium en hoog risico — op basis van hun scores. De verschillen waren opvallend: elke patiënt in de hoogrisicogroep reageerde niet op bevacizumab, terwijl de meeste patiënten in de laagrisicogroep wel een respons vertoonden. De hoogrisicogroep had ook een veel grotere kans op vroege ziekteprogressie vergeleken met de laagrisicogroep. Belangrijk is dat deze risicoscore prognostische informatie bood die verder ging dan wat artsen al konden afleiden uit standaard klinische factoren of eerdere genomische subtypering alleen.
Wat dit voor patiënten kan betekenen
Hoewel dit werk nog gevalideerd moet worden in grotere en onafhankelijke patiëntcohorten, wijst het op een toekomst waarin complexe tumor- en klinische gegevens kunnen worden geïntegreerd in één, bruikbare risicoscore. Als dit wordt bevestigd, zou een eenvoudige test die de aanwezigheid van de twee chromosomale verliezen en de BRAF-mutatie detecteert kunnen helpen om patiënten met metastatisch colorectaal carcinoom te identificeren die waarschijnlijk geen baat hebben bij combinatie‑therapie met bevacizumab. Die patiënten kunnen dan eerder naar alternatieve strategieën of klinische onderzoeken worden geleid, terwijl anderen doorgaan met een middel waarvan ze waarschijnlijker profijt hebben. Meer in het algemeen kan de hier gedemonstreerde machine learning-pijplijn worden aangepast aan andere kankersoorten en behandelingen, wat het doel van echt gepersonaliseerde kankerzorg vooruit helpt.
Bronvermelding: Thomas, V., Nyamundanda, G., Lärkeryd, A. et al. A pipeline of machine learning-driven multi-modal data fusion methods for prognostic risk analysis in bevacizumab-treated metastatic colorectal cancer. Sci Rep 16, 8843 (2026). https://doi.org/10.1038/s41598-026-39189-w
Trefwoorden: metastatisch colorectaal carcinoom, bevacizumab-resistentie, machine learning, genomische biomarkers, precisie-oncologie