Clear Sky Science · nl
scDecorr: feature-decorrelatie gebaseerde representatieleer stelt zelf-gestuurde uitlijning van meerdere single-cell-experimenten in staat
Waarom het samenbrengen van single-cell-gegevens ertoe doet
De moderne biologie kan nu de activiteit van duizenden genen in individuele cellen aflezen, waardoor zeldzame celtypen en subtiele ziektecondities zichtbaar worden. Maar deze single-cell-experimenten worden vaak in verschillende laboratoria uitgevoerd met uiteenlopende apparatuur en protocollen, wat het combineren van resultaten bemoeilijkt. Het artikel introduceert scDecorr, een nieuwe computationele methode die dergelijke diverse datasets automatisch uitlijnt zodat vergelijkbare cellen bij elkaar komen, zelfs als ze op zeer verschillende manieren zijn gemeten. Dit vereenvoudigt het bouwen van uitgebreide celatlassen en het hergebruiken van data tussen studies.
Veel datasets, één gemeenschappelijke taal
Single-cell RNA-sequencing meet welke genen in elke cel aanstaan. In theorie stelt dit wetenschappers in staat cellen te vergelijken over organen, patiënten of ziekten heen. In de praktijk kunnen technische eigenaardigheden — bekend als batch-effecten — de echte biologische verschillen overschaduwen. Celltypen kunnen verschillend lijken louter omdat ze op een andere dag of met een andere technologie zijn verwerkt. scDecorr pakt dit aan door voor elke cel een compacte numerieke "profiel" te leren, waarin cellen die zich hetzelfde gedragen dicht bij elkaar worden geplaatst, terwijl verschillende cellen uit elkaar blijven. Cruciaal is dat dit gebeurt zonder door experts opgegeven celtype-annotaties, waardoor de methode geschikt is voor grote, rommelige datasets.
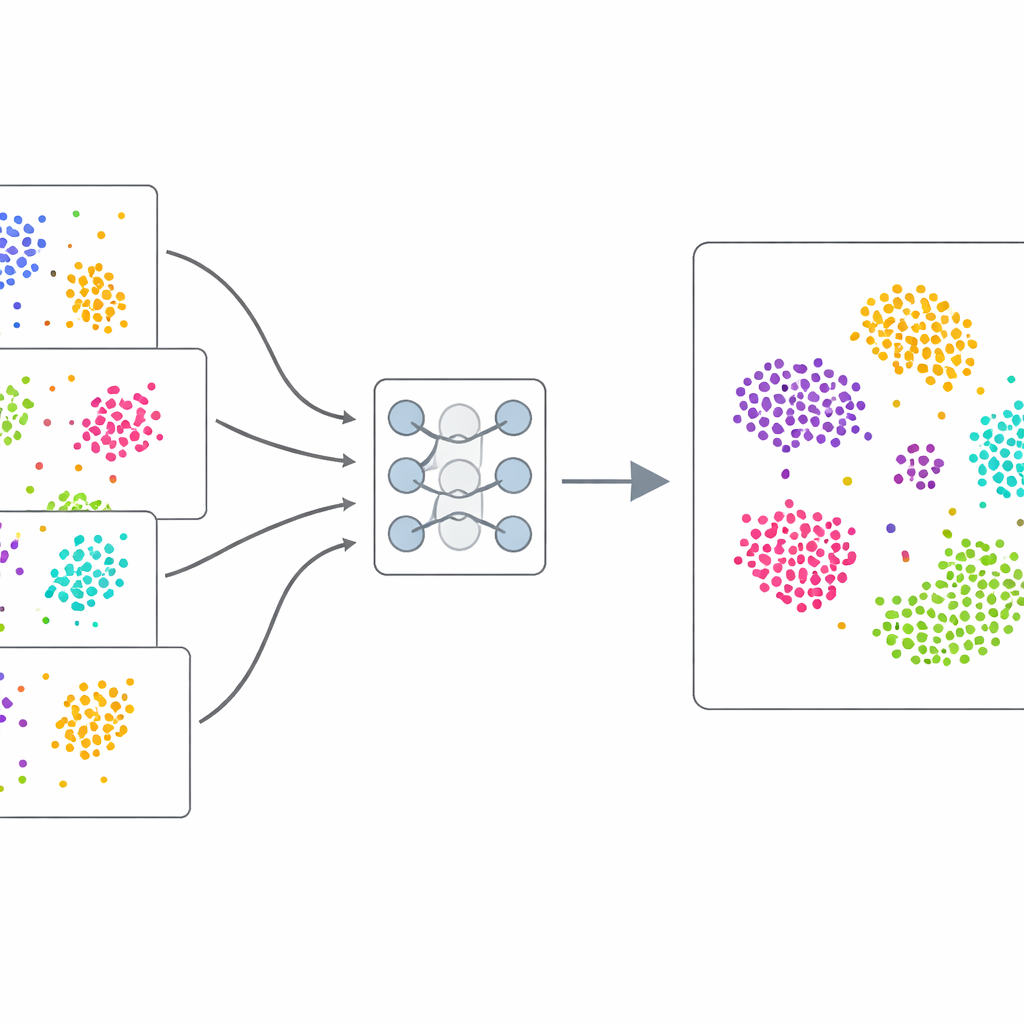
Leren van de data zelf
In plaats van te vertrouwen op handgelabelde voorbeelden, gebruikt scDecorr self-supervised learning: de data leveren hun eigen trainingssignaal. Voor elke cel maakt de methode twee licht vervormde kopieën van het genexpressiepatroon, bijvoorbeeld door willekeurig enkele waarden weg te laten of te schudden. Een tweeling-neuraal netwerk verwerkt beide versies en wordt zo getraind dat het zeer vergelijkbare interne samenvattingen produceert voor de twee weergaven van dezelfde cel, maar afwijkende samenvattingen voor verschillende cellen. Tegelijkertijd stimuleert scDecorr dat elke component van deze samenvattingen unieke informatie draagt, zodat geen enkele feature eenvoudigweg een andere dupliceert. Deze "decorrelatie"-stap voorkomt dat het model in een paar dominante patronen instort en legt in plaats daarvan een breed scala aan biologische signalen vast.
Technische verschillen subtiel corrigeren
Een centrale uitdaging is dat cellen uit verschillende studies licht verschillende statistische eigenschappen volgen. Als die naïef gemengd worden, kan het model technische verschillen als biologische signalen misinterpreteren. scDecorr pakt dit aan met een idee uit domeinadaptatie. Alle batches delen hetzelfde encoder-netwerk, maar elke batch heeft eigen normalisatielagen die features herschalen zodat binnen die batch elke dimensie een standaardvorm krijgt. Het decorrelatie-doel wordt vervolgens afzonderlijk binnen elke batch toegepast, terwijl alle batches door dezelfde encoder moeten gaan. Dit duwt de encoder zachtjes naar representaties die een gedeelde structuur over experimenten heen volgen, zodat vergelijkbare celtypen uit verschillende bronnen natuurlijk naast elkaar komen te liggen in de geleerde ruimte zonder expliciete matchingstap.
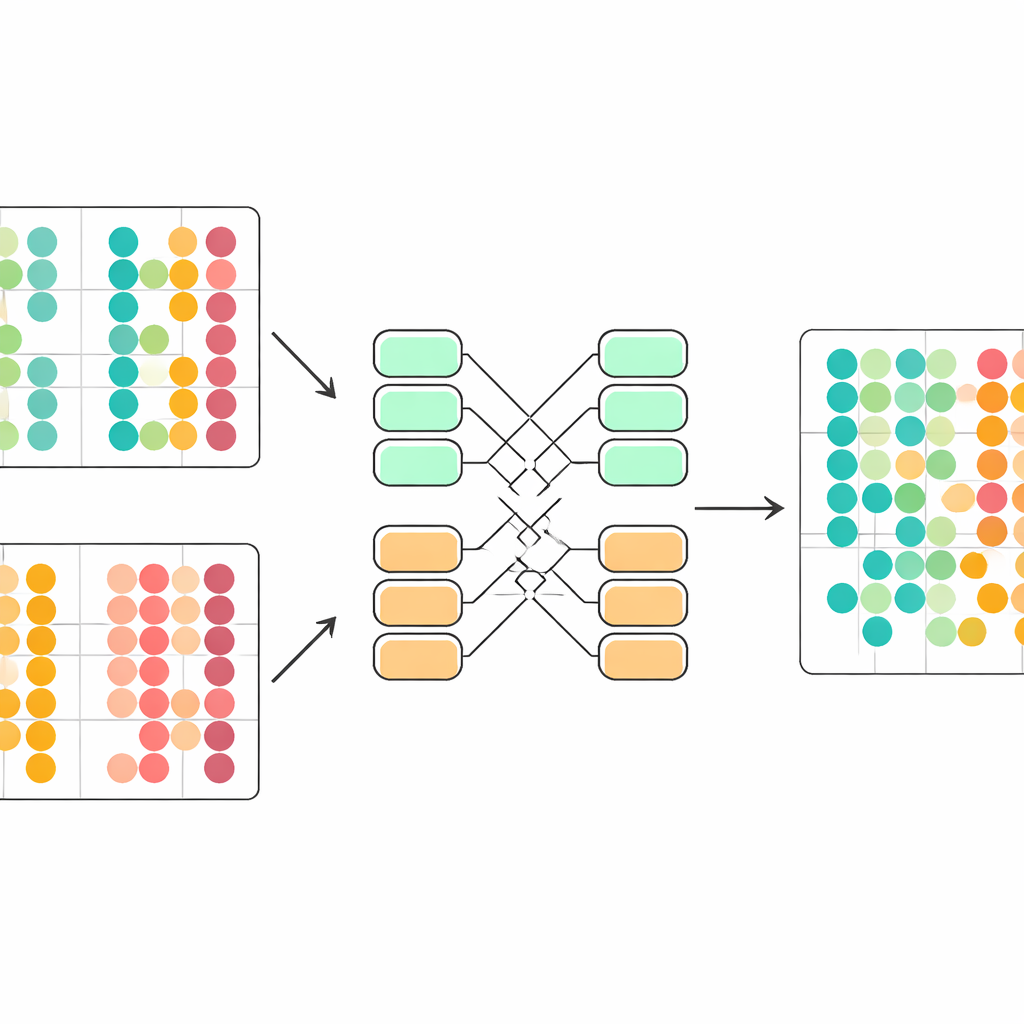
Beter dan gevestigde tools op echte datasets
De auteurs testen scDecorr grondig op vijf veeleisende verzamelingen single-cell-data, uiteenlopend over menselijke en muisweefsels, immuuncellen over organen en meerdere sequencingtechnologieën. Ze vergelijken het met verschillende veelgebruikte integratietools en met eenvoudige benaderingen zoals principale componentenanalyse. ScDecorr behoudt in taak na taak de echte biologische groeperingen van cellen beter — gemeten met standaard clustering-scores — terwijl het toch batches voldoende mengt om duidelijke technische scheidingen te verwijderen. Het is bijzonder sterk in het vermijden van overcorrectie, waarbij verschillende celtypen ten onrechte samengevoegd worden in naam van batchverwijdering, en het behoudt doorgaans duidelijke grenzen voor zeldzame of batch-specifieke celtypen die andere methoden vervagen of verliezen.
Betrouwbare overdracht van celannotaties
Naast het samenvoegen van datasets wordt scDecorr getest op labeloverdracht: het gebruiken van een geannoteerde referentiedataset om celtype-annotaties toe te kennen aan een nieuwe, niet-geannoteerde dataset. Met eenvoudige classifiers of clustering in de scDecorr-ruimte herstelt de methode betrouwbaar bekende celtypen over verschillende chemieën, platforms en studies. Het overtreft vaak of evenaart de beste bestaande tools qua classificatienauwkeurigheid, terwijl het consistenter de interne celtypestructuur binnen elke dataset bewaart. Deze prestatie blijft bestaan zelfs wanneer slechts sommige celtypen gedeeld zijn tussen datasets of wanneer batches sterk ongebalanceerd zijn, hoewel de auteurs opmerken dat extreem mismatchende instellingen voor alle methoden uitdagend blijven.
Wat dit betekent voor toekomstige celatlassen
Simpel gezegd biedt scDecorr een manier om diverse single-cell-experimenten "dezelfde taal te laten spreken" zonder zware correcties die belangrijke verschillen uitwissen. Door rijke, laag-dimensionale samenvattingen te leren die robuust zijn tegen ruis maar gevoelig voor echte biologische diversiteit, maakt het het eenvoudiger gecombineerde kaarten van cellen over weefsels, technologieën en studies te bouwen en bestaande data te hergebruiken om nieuwe experimenten te annoteren. Hoewel er ruimte is voor toekomstige verfijningen — vooral voor zeer ongebalanceerde datasets — levert scDecorr een krachtige en voorzichtige alternatieve aanpak voor batchcorrectie die wetenschappers helpt het ware cellulaire landschap met minder technische vervormingen te zien.
Bronvermelding: Sanyal, R., Xu, Y., Kim, H. et al. scDecorr: feature decorrelation based representation learning enables self-supervised alignment of multiple single-cell experiments. Sci Rep 16, 13782 (2026). https://doi.org/10.1038/s41598-026-50586-z
Trefwoorden: single-cell RNA-sequencing, data-integratie, self-supervised learning, batch-effectcorrectie, celatlas