Clear Sky Science · en
scDecorr: feature decorrelation based representation learning enables self-supervised alignment of multiple single-cell experiments
Why Bringing Single-Cell Data Together Matters
Modern biology can now read the activity of thousands of genes in individual cells, revealing rare cell types and subtle disease states. But these single-cell experiments are often done in different labs, using different machines and protocols, which makes their results hard to combine. The paper introduces scDecorr, a new computational method that automatically lines up such diverse datasets so that similar cells end up together, even if they were measured in very different ways. This makes it easier for researchers to build rich cell atlases and to reuse data across studies.
Many Datasets, One Common Language
Single-cell RNA sequencing measures which genes are switched on in each cell. In principle, this lets scientists compare cells across organs, patients, or diseases. In practice, technical quirks—known as batch effects—can drown out the true biological differences. Cells of the same type may look different just because they were processed on another day or by another technology. scDecorr tackles this by learning a compact numerical "profile" for each cell, in which cells that act alike are placed close together, while dissimilar cells are kept apart. Crucially, it does this without needing expert-provided cell-type labels, making it suitable for large, messy datasets.
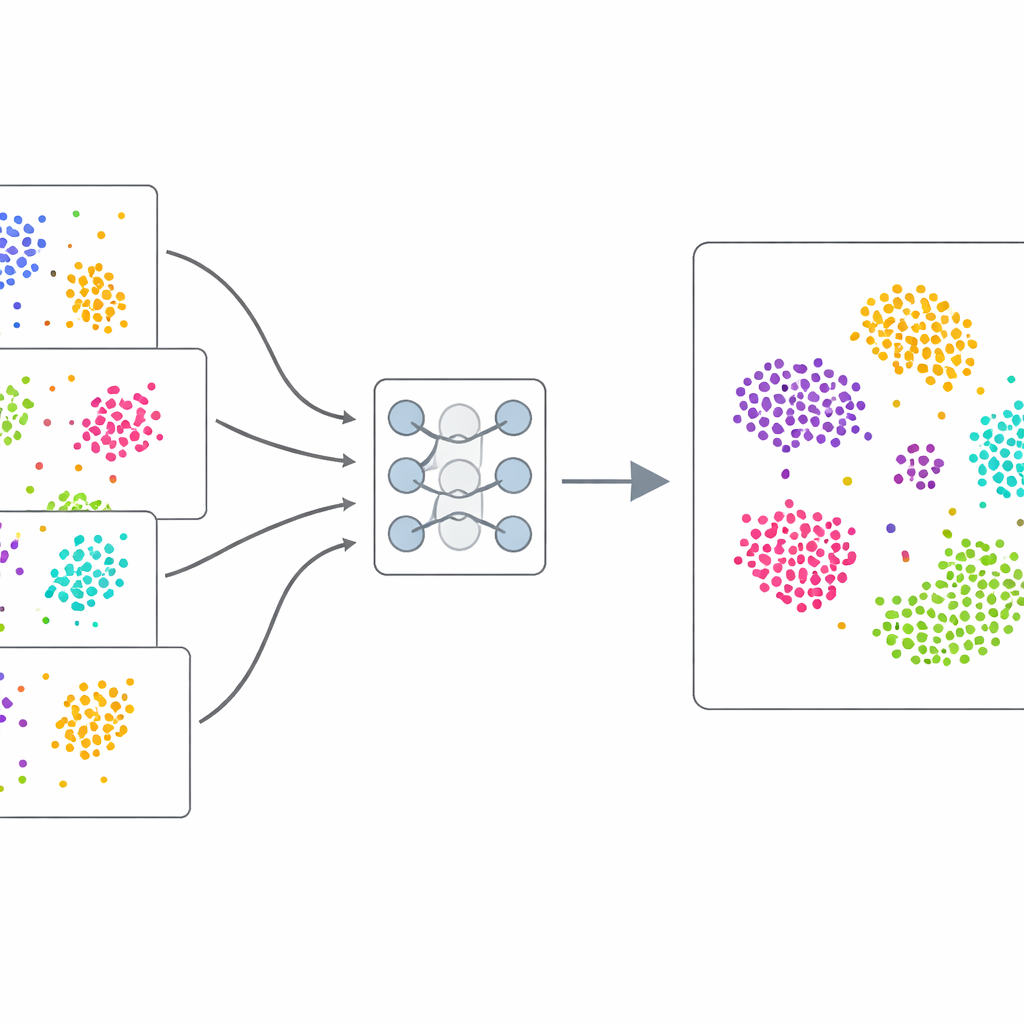
Learning From the Data Itself
Instead of relying on hand-labeled examples, scDecorr uses self-supervised learning: the data provide their own training signal. For each cell, the method creates two slightly distorted copies of its gene-expression pattern, for example by randomly dropping or shuffling some values. A twin neural network processes both versions and is trained to produce very similar internal summaries for the two views of the same cell, but dissimilar summaries for different cells. At the same time, scDecorr encourages each component of these summaries to carry unique information, so that no single feature simply duplicates another. This "decorrelation" step helps prevent the model from collapsing onto a few dominant patterns and instead captures a broad range of biological signals.
Quietly Correcting Technical Differences
A central challenge is that cells from different studies follow slightly different statistical rules. If these get mixed together naïvely, the model can misinterpret technical differences as biology. scDecorr addresses this with an idea borrowed from domain adaptation. All batches share the same encoder network, but each batch has its own normalization layers that rescale features so that, within that batch, each dimension has a standard shape. The decorrelation objective is then applied separately inside each batch, yet all batches must pass through the same encoder. This gently pushes the encoder to produce representations that follow a shared structure across experiments, so that similar cell types from different sources naturally line up in the learned space without any explicit matching step.
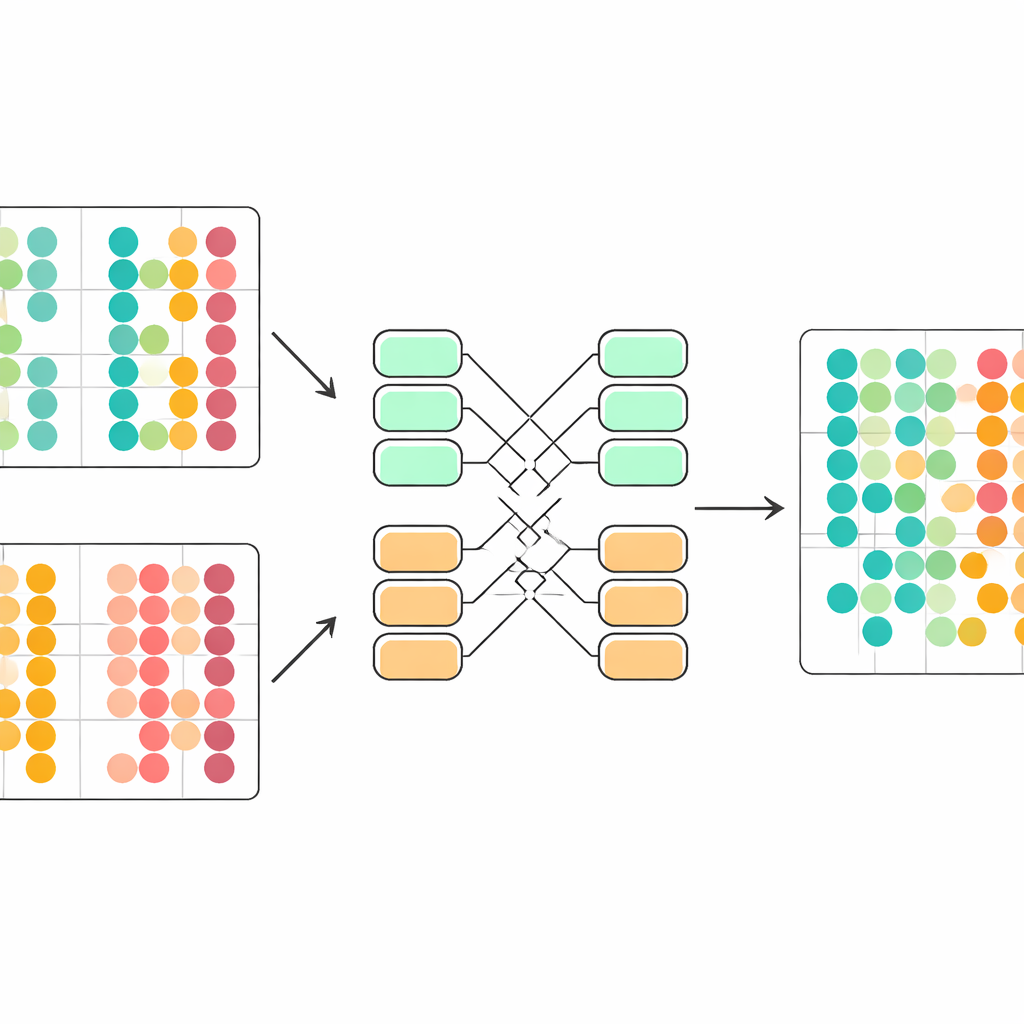
Beating Established Tools on Real Datasets
The authors rigorously test scDecorr on five demanding collections of single-cell data, spanning human and mouse tissues, immune cells across organs, and multiple sequencing technologies. They compare it with several widely used integration tools, as well as simple approaches like principal component analysis. In task after task, scDecorr better preserves the true biological groupings of cells—measured by standard clustering scores—while still mixing batches enough to remove obvious technical separation. It is especially strong at avoiding overcorrection, where different cell types get wrongly merged in the name of batch removal, and it tends to maintain clear boundaries for rare or batch-specific cell types that other methods blur or lose.
Reliable Transfer of Cell Labels
Beyond merging datasets, scDecorr is tested on label transfer: using an annotated reference dataset to assign cell-type labels to a new, unlabeled one. Using simple classifiers or clustering in the scDecorr space, the method reliably recovers known cell types across different chemistries, platforms, and studies. It often surpasses or matches the best existing tools in classification accuracy, while more consistently preserving the internal cell-type structure within each dataset. This performance holds even when only some cell types are shared between datasets, or when batches are highly unbalanced, though the authors note that extremely mismatched settings remain challenging for all methods.
What This Means for Future Cell Atlases
In plain terms, scDecorr offers a way to let diverse single-cell experiments "speak the same language" without heavy-handed corrections that erase important differences. By learning rich, low-dimensional summaries that are robust to noise yet sensitive to genuine biological diversity, it makes it easier to build combined maps of cells across tissues, technologies, and studies, and to reuse existing data to annotate new experiments. While there is room for future refinements—especially for very unbalanced datasets—scDecorr provides a powerful and more cautious alternative to batch correction, helping scientists see the true cellular landscape with fewer technical distortions.
Citation: Sanyal, R., Xu, Y., Kim, H. et al. scDecorr: feature decorrelation based representation learning enables self-supervised alignment of multiple single-cell experiments. Sci Rep 16, 13782 (2026). https://doi.org/10.1038/s41598-026-50586-z
Keywords: single-cell RNA sequencing, data integration, self-supervised learning, batch effect correction, cell atlas