Clear Sky Science · de
scDecorr: merkmalsentkoppelungsbasierte Repräsentationslerntechnik ermöglicht selbstüberwachte Ausrichtung mehrerer Einzelzell-Experimente
Warum das Zusammenführen von Einzelzell-Daten wichtig ist
Die moderne Biologie kann inzwischen die Aktivität von Tausenden Genen in einzelnen Zellen messen und damit seltene Zelltypen und feine Krankheitszustände aufdecken. Solche Einzelzell-Experimente werden jedoch oft in verschiedenen Laboren mit unterschiedlichen Geräten und Protokollen durchgeführt, wodurch sich ihre Ergebnisse schwer kombinieren lassen. Die Arbeit stellt scDecorr vor, eine neue Rechenmethode, die solche heterogenen Datensätze automatisch so ausrichtet, dass ähnliche Zellen zusammengeführt werden, selbst wenn sie sehr unterschiedlich gemessen wurden. Das erleichtert Forschenden den Aufbau umfassender Zellatlanten und die Wiederverwendung von Daten aus verschiedenen Studien.
Viele Datensätze, eine gemeinsame Sprache
Die Einzelzell-RNA-Sequenzierung misst, welche Gene in jeder Zelle an- oder abgeschaltet sind. Grundsätzlich erlaubt das den Vergleich von Zellen über Organe, Patientinnen und Patienten oder Krankheitszustände hinweg. In der Praxis können technische Abweichungen – sogenannte Batch-Effekte – die biologischen Unterschiede überlagern. Zellen desselben Typs können unterschiedlich erscheinen, nur weil sie an einem anderen Tag oder mit einer anderen Technologie verarbeitet wurden. scDecorr begegnet diesem Problem, indem es für jede Zelle ein kompaktes numerisches «Profil» lernt, in dem ähnliche Zellen nahe beieinander liegen und unähnliche auseinander. Entscheidend ist, dass dies ohne expertenbereitgestellte Zelltyp-Labels geschieht, wodurch die Methode für große, unübersichtliche Datensätze geeignet ist.
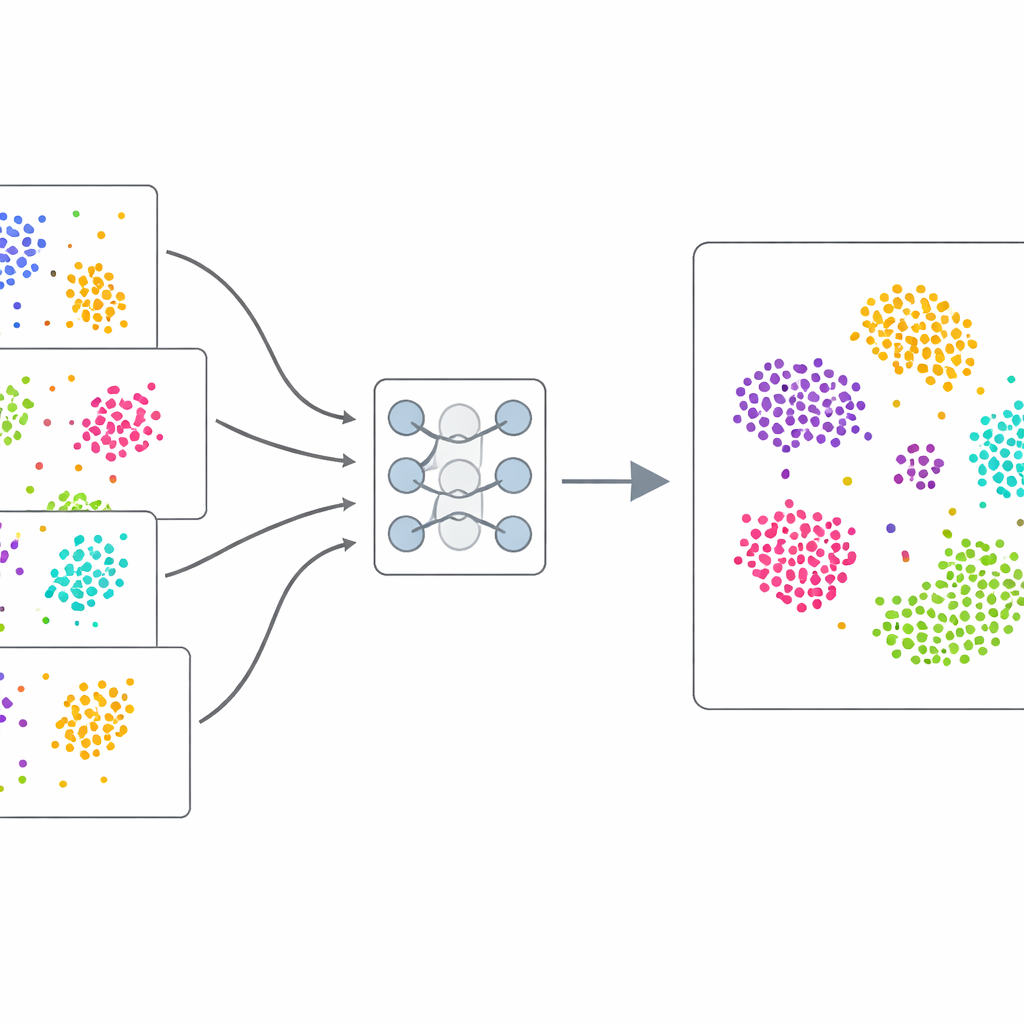
Vom Datensatz selbst lernen
Anstatt sich auf manuell gelabelte Beispiele zu stützen, verwendet scDecorr selbstüberwachtes Lernen: die Daten liefern ihr eigenes Trainingssignal. Für jede Zelle erzeugt die Methode zwei leicht veränderte Kopien ihres Genexpressionsmusters, etwa durch zufälliges Entfernen oder Vertauschen einiger Werte. Ein Zwillings-Neuronales Netz verarbeitet beide Versionen und wird so trainiert, dass es sehr ähnliche interne Zusammenfassungen für die beiden Ansichten derselben Zelle liefert, aber unterschiedliche Zusammenfassungen für verschiedene Zellen. Gleichzeitig sorgt scDecorr dafür, dass jede Komponente dieser Zusammenfassungen einzigartige Informationen trägt, sodass kein Merkmal ein anderes bloß dupliziert. Dieser Schritt der «Entkoppelung» hilft, ein Zusammenfallen auf wenige dominante Muster zu verhindern und stattdessen ein breites Spektrum biologischer Signale abzubilden.
Technische Unterschiede dezent korrigieren
Eine zentrale Herausforderung ist, dass Zellen aus verschiedenen Studien leicht unterschiedlichen statistischen Regeln folgen. Werden diese naiv zusammengeführt, kann das Modell technische Unterschiede fälschlich als biologische interpretieren. scDecorr begegnet dem mit einer Idee aus der Domänenanpassung. Alle Batches nutzen denselben Encoder, aber jeder Batch hat eigene Normalisierungsschichten, die Merkmale so umskalieren, dass innerhalb dieses Batches jede Dimension eine vergleichbare Form erhält. Das Entkoppelungs-Objektiv wird dann getrennt innerhalb jedes Batches angewendet, während alle Batches denselben Encoder passieren. Dadurch wird der Encoder behutsam dazu gedrängt, Repräsentationen zu erzeugen, die über Experimente hinweg einer gemeinsamen Struktur folgen, sodass ähnliche Zelltypen aus verschiedenen Quellen im gelernten Raum ohne expliziten Abgleich zusammenfinden.
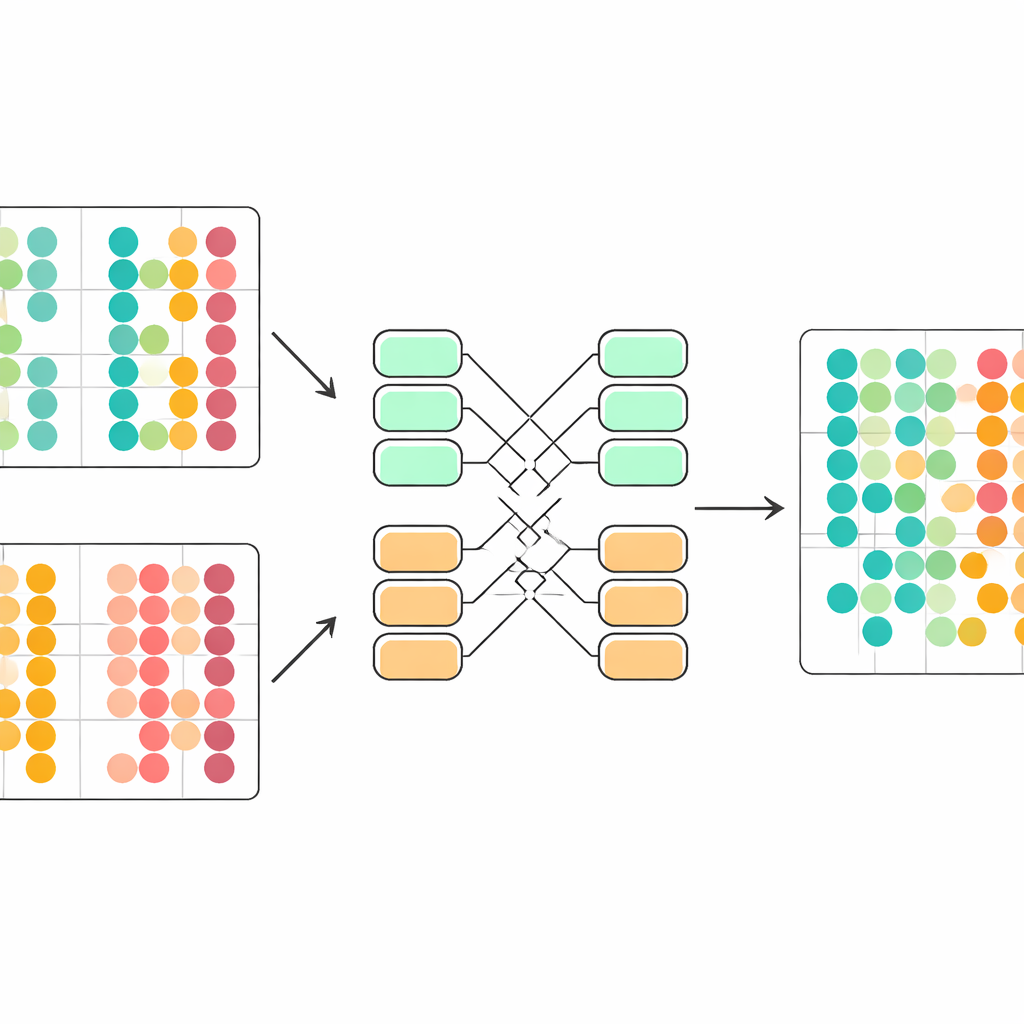
Schlägt etablierte Werkzeuge in realen Datensätzen
Die Autoren testen scDecorr gründlich an fünf anspruchsvollen Sammlungen von Einzelzelldaten, die menschliche und Maus-Gewebe, Immunzellen aus verschiedenen Organen und mehrere Sequenziertechnologien umfassen. Sie vergleichen die Methode mit mehreren weit verbreiteten Integrationswerkzeugen sowie einfachen Ansätzen wie der Hauptkomponentenanalyse. In zahlreichen Aufgaben erhält scDecorr bessere Ergebnisse bei der Erhaltung der tatsächlichen biologischen Gruppen von Zellen – gemessen mit gängigen Clustering-Maßen – und mischt gleichzeitig die Batches ausreichend, um offensichtliche technische Trennungen zu entfernen. Besonders stark ist es darin, Überkorrekturen zu vermeiden, bei denen unterschiedliche Zelltypen fälschlich zusammengeführt werden, und dabei klare Grenzen für seltene oder batch-spezifische Zelltypen zu erhalten, die andere Methoden verwischen oder verlieren.
Zuverlässige Übertragung von Zell-Labels
Über das Zusammenführen von Datensätzen hinaus wird scDecorr bei der Label-Übertragung geprüft: Mit einem annotierten Referenzdatensatz Zelltyp-Labels einem neuen, unlabeled Datensatz zuzuweisen. Mittels einfacher Klassifikatoren oder Clustering im scDecorr-Raum rekonstruiert die Methode zuverlässig bekannte Zelltypen über verschiedene Chemien, Plattformen und Studien hinweg. Häufig übertrifft oder erreicht sie die Genauigkeit der besten existierenden Werkzeuge, während sie gleichzeitig die interne Zelltyp-Struktur innerhalb jedes Datensatzes konstanter bewahrt. Diese Leistung bleibt erhalten, auch wenn nur einige Zelltypen zwischen Datensätzen geteilt werden oder die Batches stark unausgewogen sind, wenngleich die Autoren anmerken, dass extrem ungleiche Einstellungen für alle Methoden herausfordernd bleiben.
Was das für künftige Zellatlanten bedeutet
Vereinfacht gesagt bietet scDecorr eine Möglichkeit, vielfältige Einzelzell-Experimente «die gleiche Sprache sprechen» zu lassen, ohne zu starke Korrekturen, die wichtige Unterschiede auslöschen. Indem es reichhaltige, niedrigdimensionale Zusammenfassungen lernt, die gegenüber Rauschen robust und zugleich empfindlich für echte biologische Vielfalt sind, erleichtert es den Aufbau kombinierter Karten von Zellen über Gewebe, Technologien und Studien hinweg und die Wiederverwendung vorhandener Daten zur Annotation neuer Experimente. Zwar gibt es Spielraum für künftige Verbesserungen – besonders für sehr unausgewogene Datensätze – doch liefert scDecorr eine leistungsfähige und vorsichtigere Alternative zur Batch-Korrektur und hilft Forschenden, die wahre zelluläre Landschaft mit weniger technischen Verzerrungen zu erkennen.
Zitation: Sanyal, R., Xu, Y., Kim, H. et al. scDecorr: feature decorrelation based representation learning enables self-supervised alignment of multiple single-cell experiments. Sci Rep 16, 13782 (2026). https://doi.org/10.1038/s41598-026-50586-z
Schlüsselwörter: Einzelzell-RNA-Sequenzierung, Datenintegration, selbstüberwachtes Lernen, Korrektur von Batch-Effekten, Zellatlas