Clear Sky Science · it
scDecorr: apprendimento di rappresentazioni basato sulla decorrelazione delle caratteristiche consente l’allineamento self-supervised di più esperimenti single-cell
Perché è importante mettere insieme i dati single-cell
La biologia moderna è in grado di leggere l’attività di migliaia di geni in singole cellule, rivelando tipi cellulari rari e stati di malattia sottili. Tuttavia, questi esperimenti single-cell sono spesso condotti in laboratori diversi, con macchine e protocolli differenti, il che rende difficile combinare i risultati. L’articolo presenta scDecorr, un nuovo metodo computazionale che allinea automaticamente dataset eterogenei in modo che cellule simili finiscano vicine, anche se misurate con procedure molto diverse. Questo facilita per i ricercatori la costruzione di atlanti cellulari ricchi e il riutilizzo dei dati tra studi.
Molti dataset, un linguaggio comune
Il sequenziamento RNA a singola cellula misura quali geni sono attivi in ciascuna cellula. In linea di principio, ciò permette di confrontare cellule tra organi, pazienti o malattie. In pratica, però, stranezze tecniche — note come effetti batch — possono sovrastare le vere differenze biologiche. Cellule dello stesso tipo possono apparire diverse solo perché sono state processate in un giorno diverso o con una tecnologia differente. scDecorr affronta questo problema imparando un “profilo” numerico compatto per ogni cellula, in cui le cellule con comportamento simile sono collocate vicine, mentre quelle dissimili vengono mantenute distanti. Crucialmente, lo fa senza bisogno di etichette di tipo cellulare fornite da esperti, rendendolo adatto a dataset grandi e disordinati.
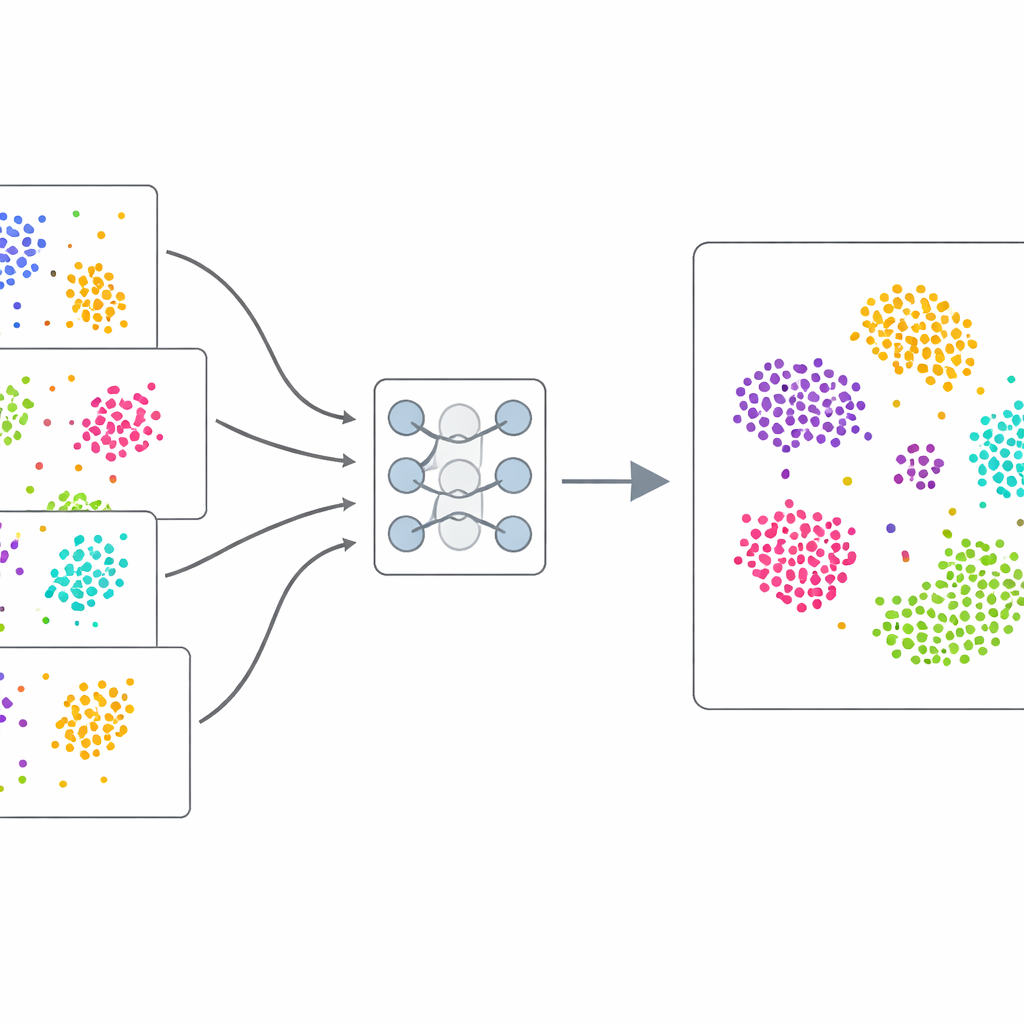
Imparare dai dati stessi
Invece di fare affidamento su esempi etichettati a mano, scDecorr utilizza l’apprendimento self-supervised: i dati forniscono il proprio segnale di addestramento. Per ogni cellula, il metodo crea due copie leggermente distorte del suo profilo di espressione genica, ad esempio cancellando o mescolando casualmente alcuni valori. Una rete neurale gemella elabora entrambe le versioni ed è addestrata a produrre sommari interni molto simili per le due viste della stessa cellula, ma sommari differenti per cellule diverse. Allo stesso tempo, scDecorr incoraggia ciascuna componente di questi sommari a portare informazioni uniche, in modo che nessuna singola caratteristica dupli semplicemente un’altra. Questo passaggio di “decorrelazione” aiuta a prevenire che il modello si concentri su pochi schemi dominanti e catturi invece una gamma ampia di segnali biologici.
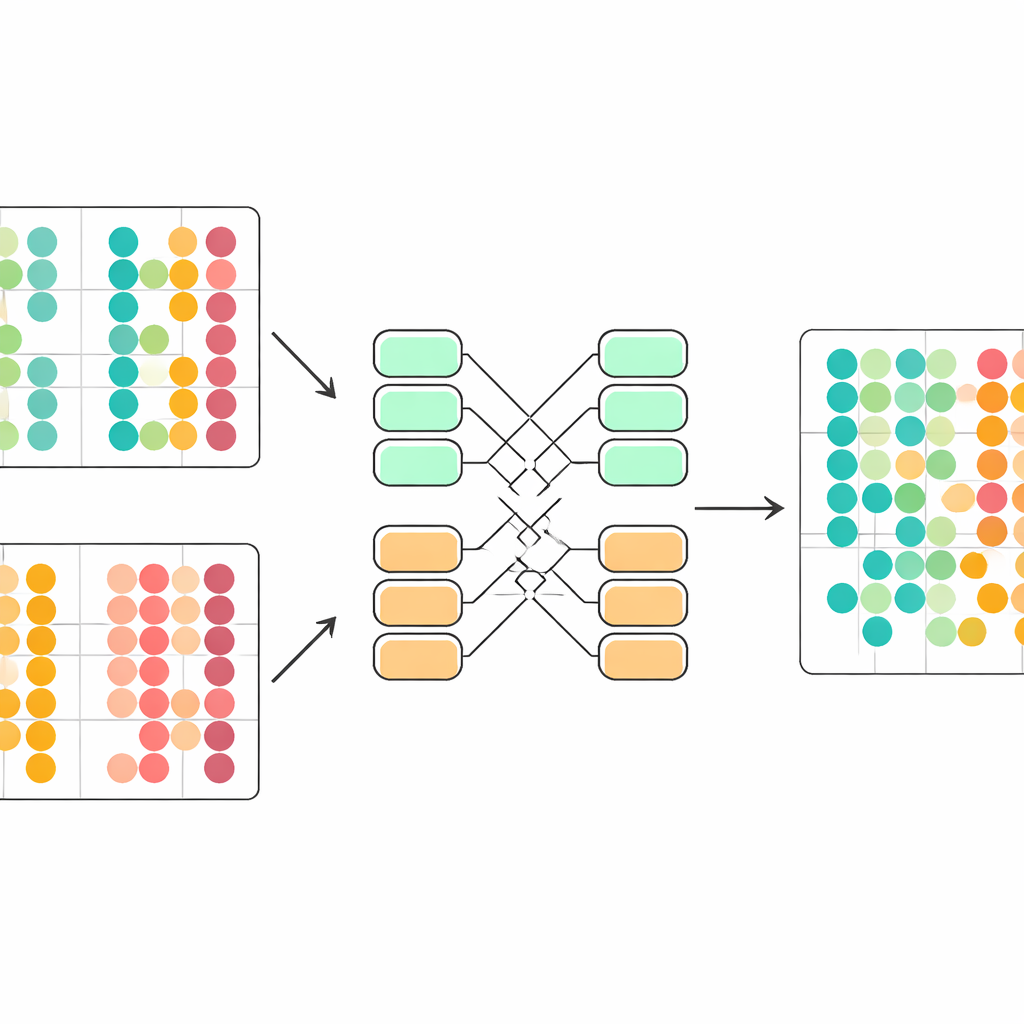
Correggere silenziosamente le differenze tecniche
Una sfida centrale è che le cellule provenienti da studi diversi seguono regole statistiche leggermente differenti. Se queste vengono mescolate alla leggera, il modello può interpretare erroneamente le differenze tecniche come biologiche. scDecorr affronta la questione con un’idea presa dall’adattamento di dominio. Tutti i batch condividono lo stesso encoder, ma ogni batch ha i propri strati di normalizzazione che riallineano le caratteristiche in modo che, all’interno di quel batch, ogni dimensione abbia una distribuzione standard. L’obiettivo di decorrelazione viene quindi applicato separatamente all’interno di ciascun batch, eppure tutti i batch passano attraverso lo stesso encoder. Questo spinge delicatamente l’encoder a produrre rappresentazioni che seguono una struttura condivisa tra gli esperimenti, così che tipi cellulari simili provenienti da fonti diverse si allineino spontaneamente nello spazio appreso senza alcun passaggio esplicito di corrispondenza.
Superare strumenti consolidati su dataset reali
Gli autori testano in modo rigoroso scDecorr su cinque raccolte impegnative di dati single-cell, che coprono tessuti umani e di topo, cellule immunitarie attraverso organi e più tecnologie di sequenziamento. Lo confrontano con diversi strumenti di integrazione largamente usati, oltre che con approcci semplici come l’analisi delle componenti principali. In compito dopo compito, scDecorr preserva meglio i veri raggruppamenti biologici delle cellule — misurati con punteggi di clustering standard — pur mescolando i batch quel tanto che basta per rimuovere la separazione tecnica evidente. È particolarmente efficace nell’evitare la sovracorrezione, quando tipi cellulari differenti vengono erroneamente fusi in nome della rimozione del batch, e tende a mantenere confini chiari per tipi cellulari rari o specifici di un batch che altri metodi sfumano o perdono.
Trasferimento affidabile delle etichette cellulari
Oltre alla fusione dei dataset, scDecorr è stato testato sul trasferimento di etichette: usare un dataset di riferimento annotato per assegnare etichette di tipo cellulare a un nuovo dataset non annotato. Utilizzando classificatori semplici o clustering nello spazio scDecorr, il metodo recupera in modo affidabile i tipi cellulari noti attraverso diverse chemistrie, piattaforme e studi. Spesso supera o eguaglia i migliori strumenti esistenti in accuratezza di classificazione, pur preservando in modo più consistente la struttura interna dei tipi cellulari all’interno di ciascun dataset. Questa performance si mantiene anche quando solo alcuni tipi cellulari sono condivisi tra i dataset, o quando i batch sono fortemente sbilanciati, anche se gli autori osservano che impostazioni estremamente non corrispondenti restano difficili per tutti i metodi.
Cosa significa questo per i futuri atlanti cellulari
In termini pratici, scDecorr offre un modo per permettere a esperimenti single-cell diversi di “parlare la stessa lingua” senza correzioni pesanti che cancellerebbero differenze importanti. Imparando riassunti ricchi e a bassa dimensione robusti al rumore ma sensibili alla vera diversità biologica, facilita la costruzione di mappe combinate di cellule attraverso tessuti, tecnologie e studi, e il riutilizzo di dati esistenti per annotare nuovi esperimenti. Pur essendoci spazio per miglioramenti futuri — specialmente per dataset molto sbilanciati — scDecorr fornisce un’alternativa potente e più cauta alla correzione del batch, aiutando gli scienziati a vedere il vero paesaggio cellulare con meno distorsioni tecniche.
Citazione: Sanyal, R., Xu, Y., Kim, H. et al. scDecorr: feature decorrelation based representation learning enables self-supervised alignment of multiple single-cell experiments. Sci Rep 16, 13782 (2026). https://doi.org/10.1038/s41598-026-50586-z
Parole chiave: sequenziamento RNA a singola cellula, integrazione dei dati, apprendimento self-supervised, correzione dell’effetto batch, atlante cellulare