Clear Sky Science · en
Extensive enhancer crosstalk controls PPARG2 activation during adipogenesis
Why Fat Cell Switches Matter
Fat cells are often blamed for obesity, but healthy fat tissue is essential for storing energy safely and keeping blood sugar and cholesterol in balance. When fat cells cannot form or function properly, fat spills into the liver, muscles, and heart, raising the risk of diabetes and heart disease. This study asks a basic but crucial question: how does one key gene get turned on at just the right time to turn an immature cell into a fully formed fat cell? By uncovering that answer, the work sheds light on why some people are more vulnerable to metabolic disease — and how tiny DNA changes far away from genes can have big effects on health.
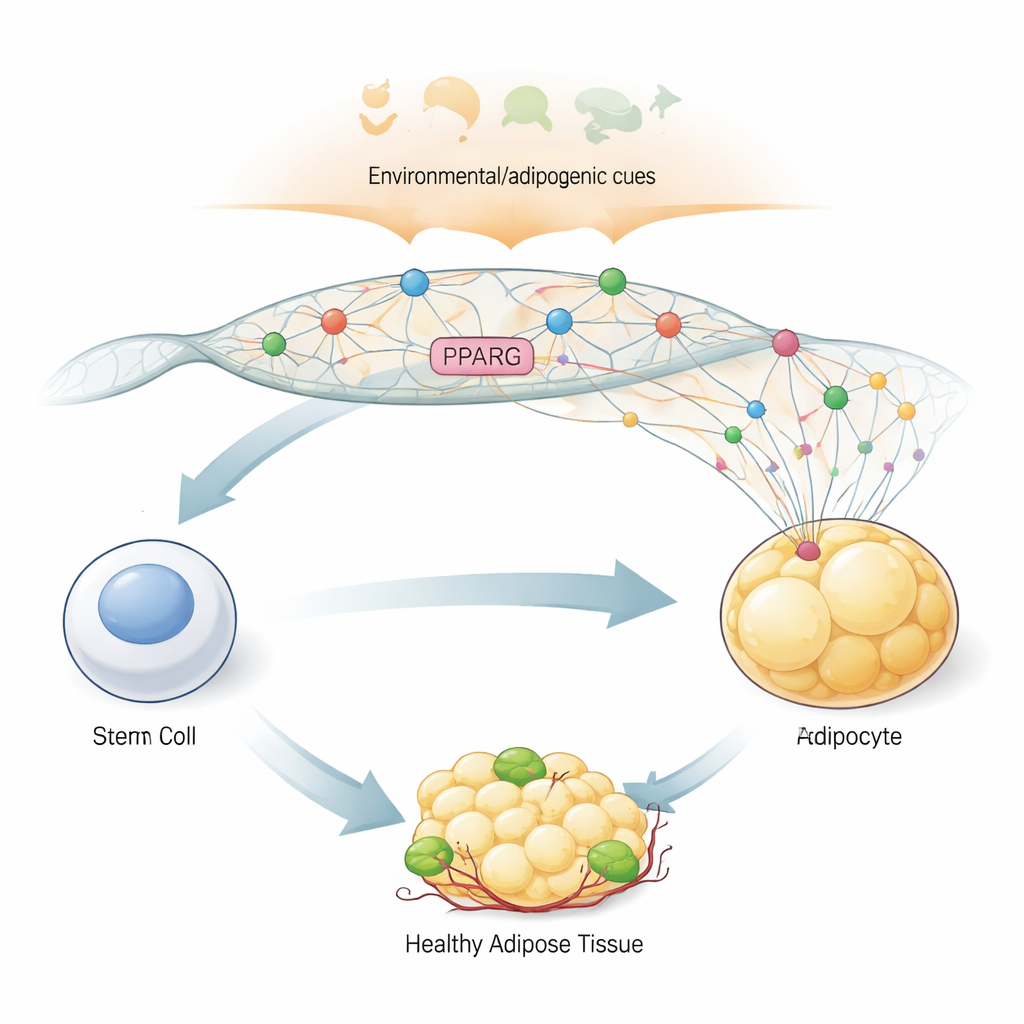
The Master Switch for Becoming a Fat Cell
To turn a generic stem-like cell into a fat cell, the body relies on a “master switch” gene called PPARG2, which produces the protein PPARγ2. This protein controls hundreds of other genes that handle fat storage, insulin sensitivity, and hormone production in fat tissue. Because of its central role, PPARG2 must stay off in precursor cells and then switch on quickly and strongly when the body sends adipogenic signals that say, in effect, “make fat cells now.” The authors used a human bone-marrow–derived stem cell model that can be steered into fat cells in the lab, allowing them to watch, step by step, how the PPARG2 region of the genome changes shape and activity as differentiation unfolds.
A 3D Neighborhood Prewired for Change
The PPARG2 gene sits inside a self-contained 3D neighborhood of DNA known as a domain, physically insulated from surrounding genes. Even before the cells commit to becoming fat, this region is already “primed”: chemical marks on the DNA and surrounding proteins flag a set of control elements, called enhancers, as future regulatory hotspots. Using 3D genome mapping methods, the researchers found that these enhancers are already talking to each other over long distances, forming a tightly wired community around PPARG2. When the cells receive adipogenic cues, these contacts strengthen, new activity marks appear, and the enhancer network focuses even more tightly on the PPARG2 promoter, coinciding with a sharp rise in PPARG2 expression.
Many Helpers, Few Spares
To test how each enhancer contributes, the team used CRISPR-based genome editing to delete nine individual enhancer segments scattered upstream, close to, and downstream of PPARG2. Rather than finding a single on/off switch, they discovered that at least six of these enhancers are essential for full PPARG2 activation and proper fat cell formation. Removing certain promoter-proximal enhancers or pieces of a large downstream “super-enhancer” greatly blunted PPARG2 activity and reduced lipid accumulation in the resulting cells. One downstream enhancer, called E+102, was especially critical: deleting it almost completely prevented PPARG2 from turning on, even though the rest of the gene and other enhancers remained intact.
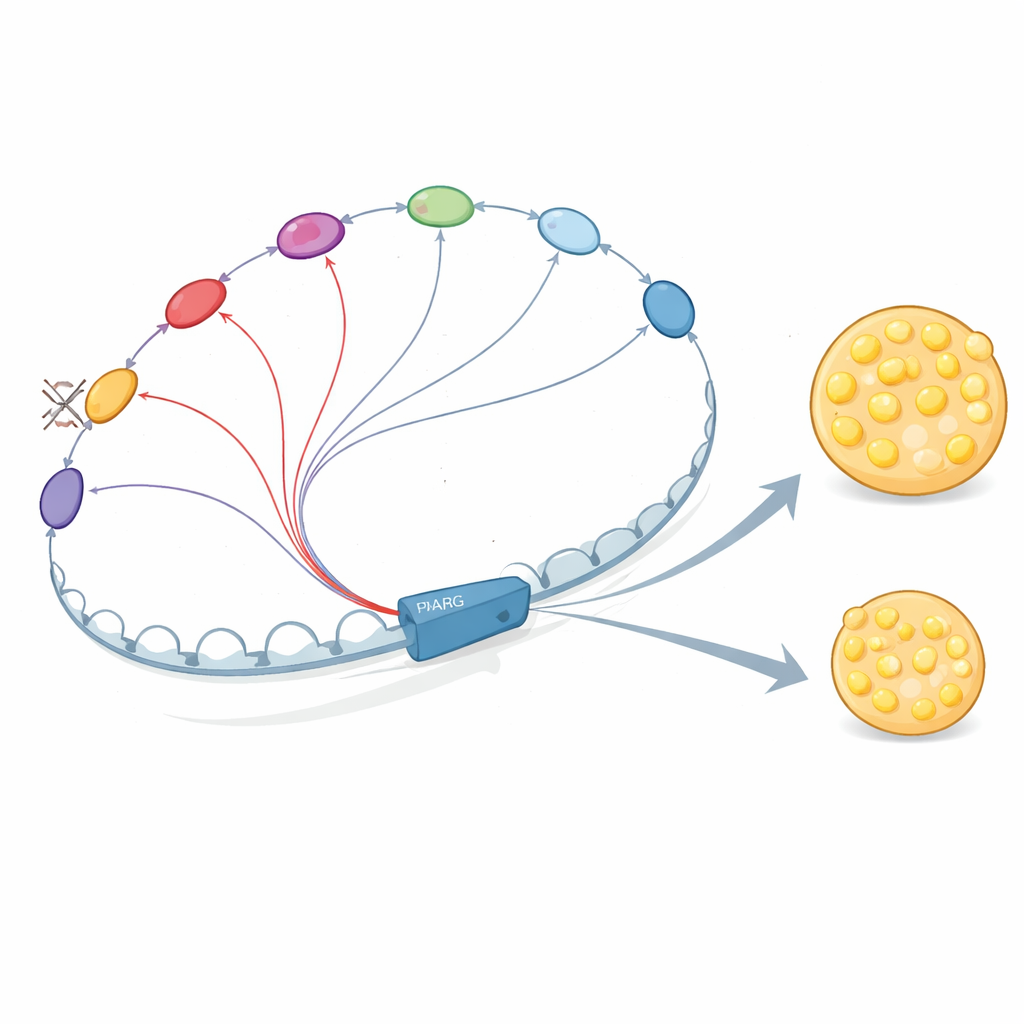
Enhancers Talking and Reinforcing Each Other
Why are these DNA elements so interdependent? The authors show that enhancers in the PPARG2 neighborhood do not act in isolation. They recruit key regulatory proteins, including early-acting factors like C/EBPβ and later, PPARγ itself and C/EBPα. These proteins bind at multiple enhancers that physically contact one another, stabilizing each other’s binding and boosting the recruitment of the Mediator complex, which helps initiate transcription. When the researchers removed E+102 or a key promoter-proximal enhancer, binding of these factors and Mediator dropped not just at the deleted site but across other enhancers in the region, and the 3D contact network weakened. This supports a picture in which enhancer–enhancer “crosstalk” creates a cooperative hub that amplifies the PPARG2 signal.
From DNA Variants to Disease Risk
Finally, the study links this intricate control system to human metabolic disease. Most DNA variants associated with obesity, cholesterol levels, or type 2 diabetes lie outside genes, in non-coding regions like enhancers. By combining large genetic datasets with a powerful machine-learning model of genome regulation, the authors found that many top-scoring risk variants within the PPARG2 domain fall directly inside the most critical enhancers they had mapped, including E+102 and promoter-proximal sites. Some of these variants are predicted — and in one case shown — to alter how well PPARγ or other factors bind, subtly changing enhancer activity and PPARG2 expression. In other words, small tweaks to this enhancer network may nudge a person’s fat cells toward healthier or less healthy behavior.
What This Means for Metabolic Health
In everyday terms, this work reveals that turning on the key fat-cell gene PPARG2 is not the job of a single switch but of a tightly choreographed team of DNA control elements that talk to each other in three dimensions. One especially important enhancer both helps start the process and then serves as a main target for positive feedback from PPARγ protein itself, ensuring a rapid and robust commitment to the fat-cell fate. Genetic variants that weaken parts of this enhancer community may lower PPARG2 activation, impair fat cell formation or function, and thereby contribute to cardiometabolic disease. Understanding this networked control system opens the door to more precise ways of assessing risk and, potentially, tuning fat cell biology to improve metabolic health.
Citation: Cetnarowska, A., Hyldahl, M., Nygård, M. et al. Extensive enhancer crosstalk controls PPARG2 activation during adipogenesis. Nat Commun 17, 3824 (2026). https://doi.org/10.1038/s41467-026-70401-7
Keywords: PPARG2, enhancers, adipogenesis, gene regulation, cardiometabolic disease