Clear Sky Science · en
A reversible allosteric inhibitor of GlyT2 for neuropathic pain without on-target side effects
New hope for stubborn nerve pain
Neuropathic pain—lingering pain caused by nerve damage—often resists standard treatments and pushes patients toward long‑term opioid use. This study introduces a new small‑molecule drug candidate that tamps down nerve‑signal traffic in the spinal cord without the heavy baggage of addiction or severe side effects. By revealing both how the drug works at the atomic level and how it behaves in animals, the researchers outline a path toward safer, non‑opioid relief for chronic nerve pain.
Why current pain drugs fall short
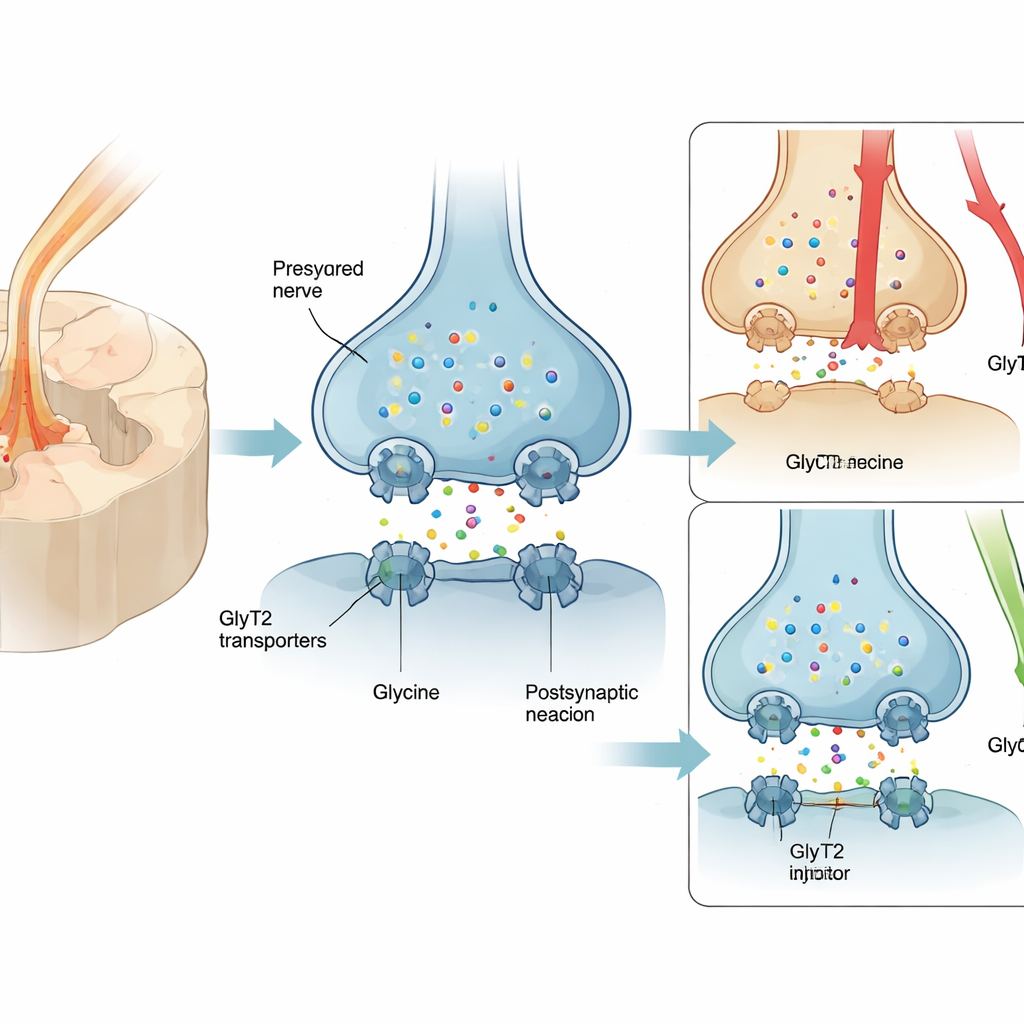
Many people with nerve pain are prescribed pregabalin or related medicines, yet only about one in four patients experience meaningful relief, and side effects such as dizziness and drowsiness are common. As a result, opioids are frequently used despite the risk of dependence. One promising alternative target is a protein called GlyT2, which sits on nerve endings in the spinal cord and recycles glycine, a calming chemical messenger that dampens pain signals. Boosting glycine around these synapses can strengthen the spinal cord’s natural “brakes” on pain. Earlier GlyT2 blockers, however, clung too tightly to the transporter and caused serious on‑target problems—tremors, seizures and even death in animals—blocking their path to the clinic.
A gentler way to nudge a key pain gate
The team designed RPI‑GLYT2‑82, a new compound built from the scaffold of a powerful older inhibitor, ORG25543, but intentionally tuned to bind less tightly and to let go more quickly. In frog egg cells engineered to produce human GlyT2, RPI‑GLYT2‑82 blocked the transporter at sub‑micromolar concentrations yet washed out within minutes, while ORG25543 remained stuck for far longer. Crucially, RPI‑GLYT2‑82 preferred GlyT2 over its close relative GlyT1, which is widespread throughout the brain, reducing the risk of widespread disruption of glycine signaling. The compound blocked transport even when glycine levels were high, confirming that it acts at a separate control site rather than directly competing with glycine itself.
Seeing the pain switch at atomic detail
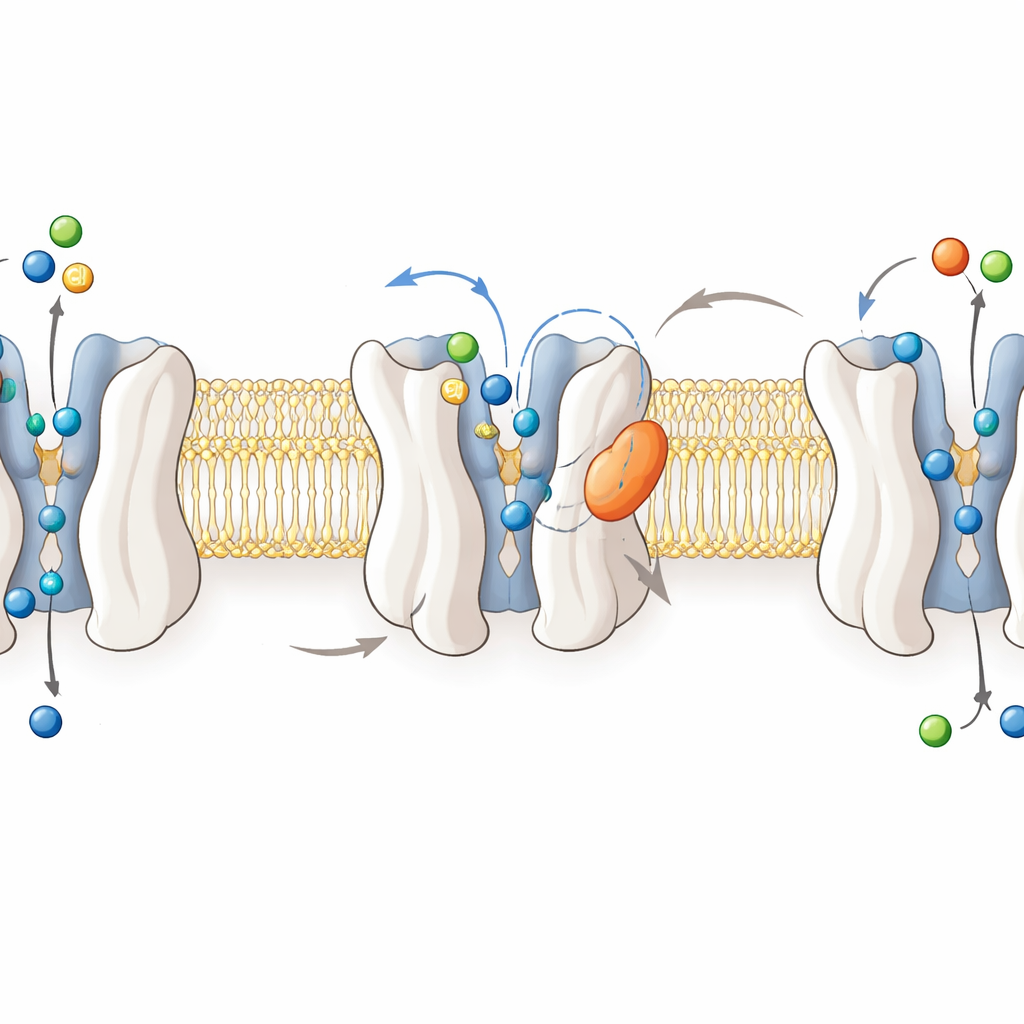
To understand how the drug works, the researchers solved high‑resolution cryo‑electron microscopy structures of human GlyT2 in four states: empty, carrying glycine, and bound to either ORG25543 or RPI‑GLYT2‑82. These images show GlyT2 as a bundle of twelve membrane‑spanning helices that rock between inward‑facing and outward‑facing shapes as they move glycine, sodium, and chloride across the cell membrane. Glycine sits deep in a central pocket when the transporter is “occluded,” shielded from both sides of the membrane. In contrast, both inhibitors lodge in an “allosteric” pocket on the outside of the protein, wedged between helices and a flexible loop. There they pry apart key gate residues and pin an important “gatekeeper” leucine outside the central pocket, freezing GlyT2 in an outward‑open, non‑transporting pose.
What makes the new molecule safer
RPI‑GLYT2‑82 and ORG25543 bind to the same allosteric site and make many of the same contacts, but subtle chemical tweaks change how they sit in the pocket. Computer simulations revealed that ORG25543 nestles deeply into a snug hydrophobic niche, with a compact ring system that fits like a plug and stabilizes the locked state. By contrast, RPI‑GLYT2‑82 carries a bulkier, more polar ring and a more strained orientation of one of its aromatic groups. These features weaken its grip, increase its motion within the pocket, and make it more likely to drift back into the surrounding solution—explaining its lower potency but much faster reversibility. Mutations in nearby amino acids that loosen these interactions further speed the release of both inhibitors, underscoring how the local geometry tunes drug residence time and, consequently, safety.
Testing pain relief and safety in animals
In mouse models where the sciatic nerve is partially damaged, animals develop mechanical and cold allodynia—pain from light touch or mild cooling. When given by injection, RPI‑GLYT2‑82 reduced both types of hypersensitivity in a dose‑dependent fashion, with peak relief appearing one to three hours after dosing. The degree of benefit was similar to that of the reference drugs gabapentin or pregabalin for certain tests, though not superior across the board. Importantly, doses that relieved pain did not impair balance, grip strength, or breathing, and even a five‑fold higher dose caused only transient, reversible motor and respiratory effects. In a conditioned place‑preference test—a standard way to probe drug reward—mice developed a clear preference for morphine, but not for RPI‑GLYT2‑82, suggesting a low risk of addiction‑like reinforcing effects.
What this means for future pain treatments
Taken together, the work shows that it is possible to dial in a GlyT2 inhibitor that enhances the body’s own inhibitory glycine signaling, relieves neuropathic pain in animals, and avoids the severe on‑target side effects that plagued earlier compounds. By providing detailed structural blueprints of GlyT2 in action and demonstrating the benefits of a reversible, allosteric approach, the study opens the door to a new class of non‑opioid painkillers. With further optimization of potency, selectivity, and pharmacokinetics, analogues of RPI‑GLYT2‑82 could eventually give patients with chronic nerve pain an effective option that is less risky than long‑term opioid therapy.
Citation: Cantwell Chater, R.P., Peiser-Oliver, J., Pati, T.K. et al. A reversible allosteric inhibitor of GlyT2 for neuropathic pain without on-target side effects. Nat Commun 17, 2828 (2026). https://doi.org/10.1038/s41467-026-69616-5
Keywords: neuropathic pain, glycine transporter, non-opioid analgesic, allosteric inhibition, cryo-EM structure