Clear Sky Science · en
Periosteal mitochondria DNA structures drive aging-associated poor skeletal repair
Why Broken Bones Struggle to Heal in Old Age
As people grow older, a simple fall can lead to a broken bone that never quite heals right. Doctors see this every day, but the root cause inside our cells has been unclear. This study uncovers a surprising culprit buried deep within the tiny power stations of bone-repair cells—unusual DNA knots that quietly build up over time and sabotage healing.
A Thin Layer with a Big Job
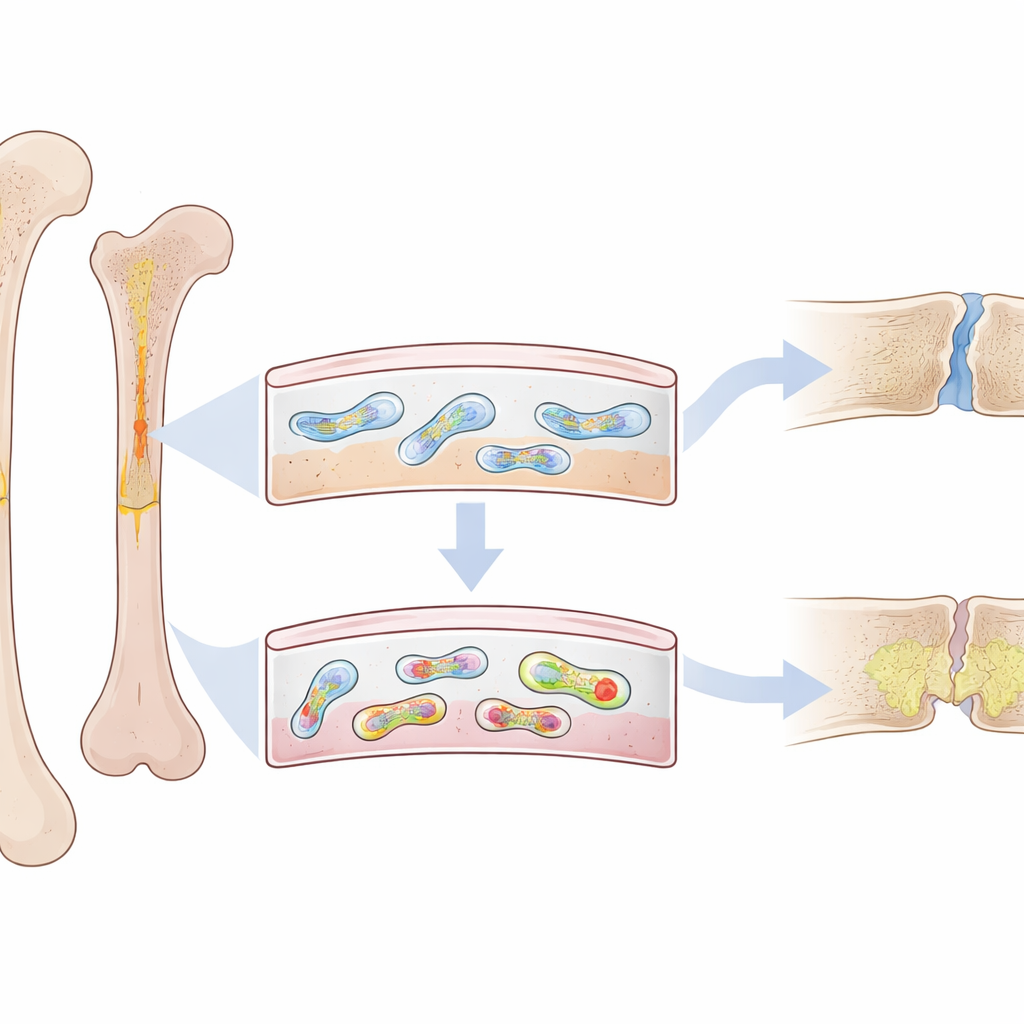
On the outside of every bone lies a thin, fibrous skin called the periosteum. Far from being just a wrapper, it holds a special group of stem-like cells that rush in to rebuild bone after a fracture. In young adults, these periosteal cells are highly active and can quickly form new, solid bone. But in older people—and in animals with premature aging—repairs are slower, weaker, and more likely to leave gaps filled with soft, fragile tissue. The researchers suspected that something inside these periosteal cells changes with age, tipping them away from healthy rebuilding.
Tiny DNA Knots in Cell Powerhouses
The team focused on mitochondria, the energy factories inside cells that carry their own small loops of DNA. Certain stretches of this mitochondrial DNA can fold into tight, four-stranded knots known as G-quadruplexes. Using a highly specific fluorescent probe, the scientists mapped these structures in mouse bones. They discovered that these DNA knots, called mtG4, pile up selectively in the periosteum as animals age—both in normal old mice and in a genetic model of premature aging. Importantly, this buildup was strongest in the periosteal stem cells that normally drive fracture repair.
From Energy Crisis to “Old” Cells
To see what mtG4 actually does, the researchers isolated periosteal stem cells and grew them into tiny 3D organoids—mini tissues that mimic early fracture repair. By briefly exposing the cells to potassium, which stabilizes G-quadruplex structures, they boosted mtG4 formation without otherwise harming the cells. The consequences were striking: key mitochondrial genes became less active, energy production (ATP) fell, and the mitochondria themselves turned swollen and damaged, often being swallowed up by recycling structures in a process called mitophagy. These stressed cells took on hallmarks of cellular old age—stopping division, expressing classic “senescence” markers, and releasing high levels of inflammatory molecules that can damage nearby tissue.
When Repair Cells Choose the Wrong Path
Healthy periosteal stem cells can choose to become hard, mineralized bone or flexible cartilage, and that balance is crucial for proper healing. In the organoid model, cells overloaded with mtG4 lost much of their ability to form bone, showing less mineral buildup and lower levels of bone-related genes. At the same time, they became more prone to forming cartilage-like tissue, with bigger, more mature cartilage clusters. When these mtG4-rich organoids were transplanted into mice, they generated far less new bone than normal organoids, faithfully mimicking the poor repair seen in aged animals.
Linking DNA Structures to Fragile Bones
Finally, the team tested whether this mechanism truly drives age-like bone problems in living animals. In mice carrying a mutation that destabilizes mitochondrial DNA and promotes mtG4 formation, fractures healed poorly: the new bone was thin, porous, and mechanically weak, while cartilage lingered where solid bone should have formed. Across these experiments, one pattern emerged—wherever mtG4 accumulated in periosteal stem cells, mitochondria failed, cells became senescent, and bone repair suffered.
What This Means for Healthy Aging
The study reveals that special DNA knots inside mitochondrial DNA can act like molecular handbrakes on bone repair, especially in the periosteum. As mtG4 structures accumulate with age, they drain energy from stem cells, push them into a prematurely “old” state, and skew healing toward soft cartilage instead of strong bone. By pinpointing mtG4 as a tissue-specific marker and driver of harmful senescent cells, this work suggests a new route for future treatments: drugs that prevent or undo these mitochondrial DNA knots, or that selectively remove mtG4-laden senescent cells, could one day help older patients heal broken bones more quickly and securely.
Citation: Wu, Y., Han, C., Yang, X. et al. Periosteal mitochondria DNA structures drive aging-associated poor skeletal repair. Bone Res 14, 40 (2026). https://doi.org/10.1038/s41413-026-00524-6
Keywords: bone repair, aging, mitochondria, stem cells, cellular senescence