Clear Sky Science · de
Deacylative Kopplung zwischen Ketonen via oxidative SH2-homolytische Substitution
Gewöhnliche Chemikalien zu flexiblen Bausteinen machen
Chemiker bauen komplexe Moleküle für Arzneimittel und Materialien mit einer kleinen Auswahl einfacher Ausgangsstoffe. Diese Studie zeigt, wie sich eines der gebräuchlichsten Bausteine, Ketone, dazu bringen lässt, neue Verbindungen zu bilden, die zuvor nur schwer zugänglich waren, und eröffnet so neue Wege, drugähnliche Moleküle schneller und präziser zu entwerfen.
Warum alltägliche Ketone wichtig sind
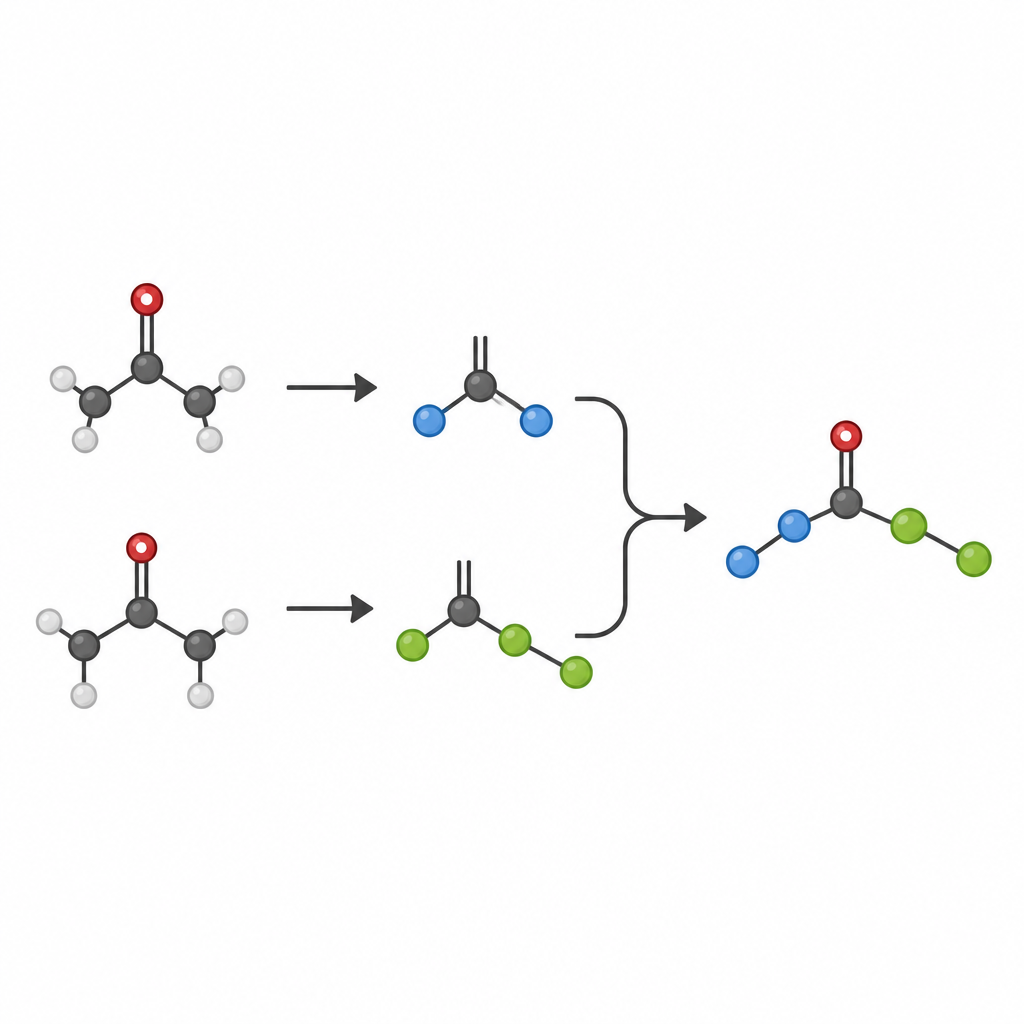
Ketone sind Arbeitspferde der organischen Chemie: geschätzt, weil sie sich leicht herstellen, lagern und modifizieren lassen. Klassische Reaktionen wie die Aldol-Reaktion oder die McMurry-Kupplung wandeln Ketone bereits in komplexere Produkte um, doch diese Methoden benötigen oft große Mengen Metallreagenzien und tun sich schwer, zwei unterschiedliche Partner kontrolliert zu vereinen. Besonders das Bilden stabiler Verbindungen zwischen zwei gesättigten Kohlenstoffatomen — die sogenannten C–C-Einfachbindungen in sterisch engen Umgebungen — blieb eine große Herausforderung, obwohl solche überfüllten Kohlenstoffzentren zentrale Merkmale vieler moderner Arzneimittel sind.
Ein neuer Weg, zwei Radikale zusammenarbeiten zu lassen
Die Autoren wollten erreichen, dass zwei verschiedene Ketone auf cleane und selektive Weise miteinander reagieren. Statt die übliche Carbonylgruppe des Ketons zu aktivieren, konzentrieren sie sich darauf, in der Nähe liegende C–C-Bindungen zu spalten, um kleine, kurzlebige Fragmente freizusetzen, die als Radikale bezeichnet werden. Diese Radikale können prinzipiell auf vielfältige Weise wieder miteinander reagieren, einschließlich unerwünschter Selbstpaarung oder Nebenreaktionen. Um dieses Chaos zu zähmen, verwendet das Team eine Strategie namens SH2, eine Form der homolytischen Substitution, bei der ein Metallkomplex ein Radikal kurzfristig bindet und dann von einem zweiten Radikal von außen angegriffen wird. Dieser Außen-Sphären-Weg hilft zu steuern, welche zwei Teile tatsächlich zusammenkommen.
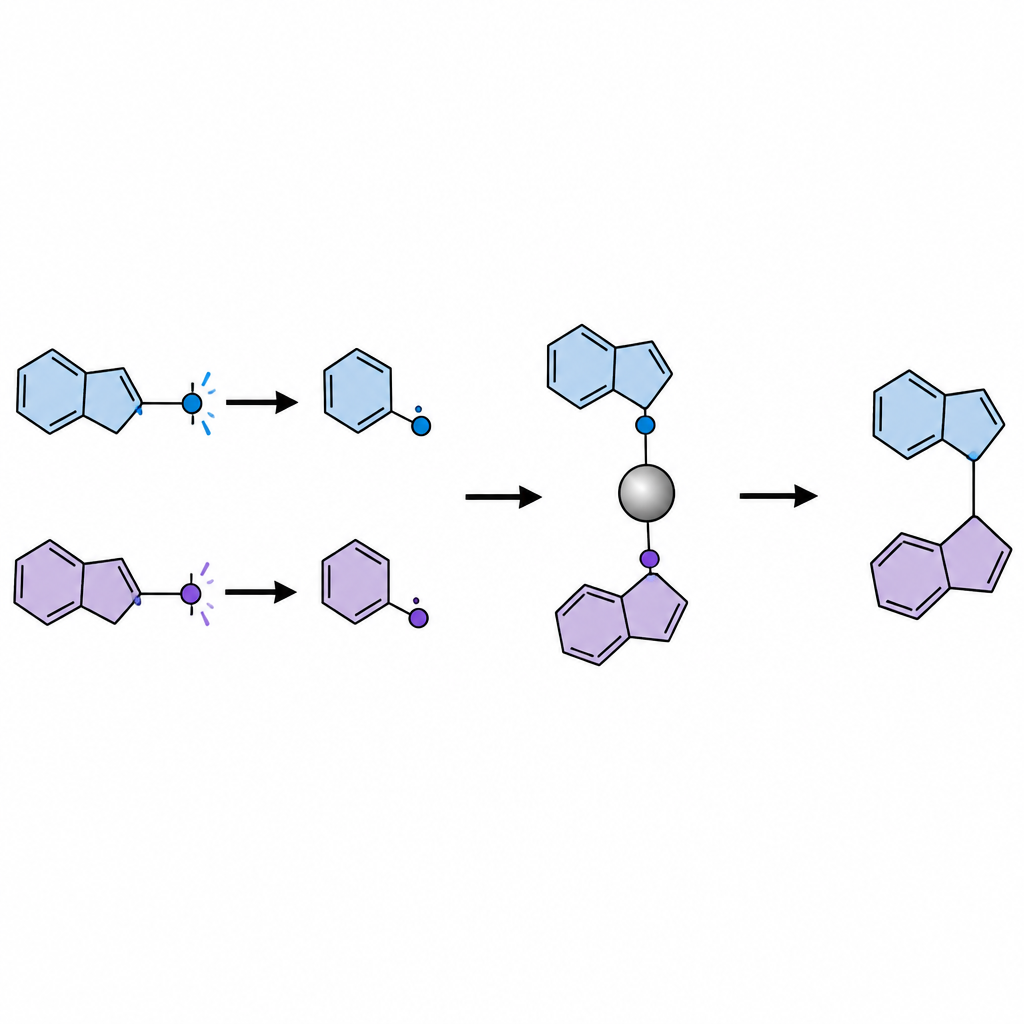
Ketone maskieren, um ihr Verhalten zu kontrollieren
Direktes Belichten von Ketonen erzeugt oft nützliche und schädliche Radikalfragmente zugleich. Um dieses Problem zu umgehen, wandeln die Forscher jedes Keton in einer einfachen Schrittfolge in eine bankstabile, „maskierte“ Form namens Dihydroquinazolinon um, bevor die Hauptreaktion startet. Unter blauem Licht oxidiert ein organischer Photokatalysator diese maskierten Ketone, wodurch sie auseinanderbrechen und zwei unterschiedliche Radikale freisetzen: ein kleineres, weniger gehindertes primäres Fragment und ein größeres sekundäres oder tertiäres Fragment. Ein Nickel-Katalysator ergreift selektiv das kleinere Radikal und bildet einen persistierenden Nickel–Kohlenstoff-Komplex, während das voluminösere Radikal frei in Lösung verbleibt. Da die Bindung des Nickels an das primäre Radikal stärker ausfällt als an ein tertiäres Radikal, sortiert das System die Radikale auf natürliche Weise in ihre unterschiedlichen Rollen.
Aufbau sterisch gehinderter Zentren für drugähnliche Moleküle
Sobald der Nickel–primär-Komplex gebildet ist, wird er in einem SH2-Schritt vom größeren Radikal angegriffen, wodurch eine neue C–C-Bindung entsteht und der Nickel-Katalysator regeneriert wird. Mit diesem Ansatz stellt das Team eine breite Palette von Produkten her, die ein sterisch gehindertes, vierfach substituiertes Kohlenstoffzentrum neben einem Stickstoff-, Sauerstoff- oder Schwefelatom enthalten. Dazu zählen β‑quaternäre Amine, β‑Aminoalkohole, β‑Diamine, β‑Aminothiole und verwandte Ether—alles Motive, die in bioaktiven Verbindungen häufig vorkommen. Die Reaktion verträgt viele funktionelle Gruppen wie Alkene, Ester, Halogenide und Ringsysteme und läuft unter milden Bedingungen mit sichtbarem Licht. Die Autoren zeigen außerdem, dass die Methode spät in der Synthese bekannter Wirkstoffmoleküle angewendet werden kann, wobei deren sensible Bereiche schonend erhalten bleiben.
Ein Blick unter die Haube der Reaktion
Um den Ablauf zu verstehen, fangen die Forscher die flüchtigen Radikale ein, untersuchen, wie der lichtabsorbierende Katalysator gequencht wird, und berechnen die relativen Geschwindigkeiten und Energien wichtiger Schritte. Ihre Befunde deuten auf einen Mechanismus hin, bei dem beide Radikale aus der Oxidation der maskierten Ketone entstehen und nicht aus dem Peroxid‑Oxidationsmittel, und bei dem Nickel in einem höheren Oxidationszustand verbleibt, wodurch der typische Innen‑Sphären‑Kreuzkupplungsweg mit Arylhalogeniden vermieden wird. Berechnungen zeigen, dass die Bildung des Nickel–primär-Komplexes energetisch bevorzugt ist und dass beide Radikale mit ähnlichen Raten erzeugt werden — Bedingungen, die unerwünschte Selbstkupplung unterdrücken und die gewünschte Kreuzpaarung begünstigen.
Was das für die zukünftige Molekülherstellung bedeutet
Einfach gesagt, lehrt diese Arbeit Ketone einen neuen Trick: sich kontrolliert aufzubrechen, sodass die richtigen Fragmente wieder zu neuen, komplexeren Strukturen zusammengesetzt werden. Durch die Kombination von sichtbarem Licht, einem Nickelkatalysator und geschickt maskierten Ketonen schaffen die Autoren eine allgemeine und flexible Route zu sterisch gehinderten Kohlenstoffzentren, die in der Wirkstoffforschung hoch geschätzt werden. Diese Strategie erweitert das Werkzeugarsenal der Chemiker zum Bearbeiten und Diversifizieren von Molekülen, insbesondere in späten Synthesestufen, in denen kleine Änderungen große Auswirkungen auf die biologische Aktivität haben können.
Zitation: Yang, JX., Zhang, MY., Wen, Q. et al. Cross-ketone deacylative coupling via oxidative SH2 homolytic substitution. Nat Commun 17, 4248 (2026). https://doi.org/10.1038/s41467-026-70619-5
Schlüsselwörter: Ketonkopplung, radikalische Chemie, Nickelkatalyse, Photoredox, quaternäres Kohlenstoff