Clear Sky Science · zh
通过自回归等变网络实现无力分子动力学
为何加速原子“电影”至关重要
化学、物理和材料科学中许多最引人入胜的问题归根结底都与原子的运动有关:止痛药分子如何摆动,石英如何振动,水如何冻结成奇特的玻璃态。名为分子动力学的计算实验模拟这些原子级电影,但它们计算量极大,科研人员通常只能看到微小一部分过程。本文介绍了 TrajCast,一种新的人工智能框架,它学会在不牺牲关键物理特性的前提下,将这些原子影片快进。
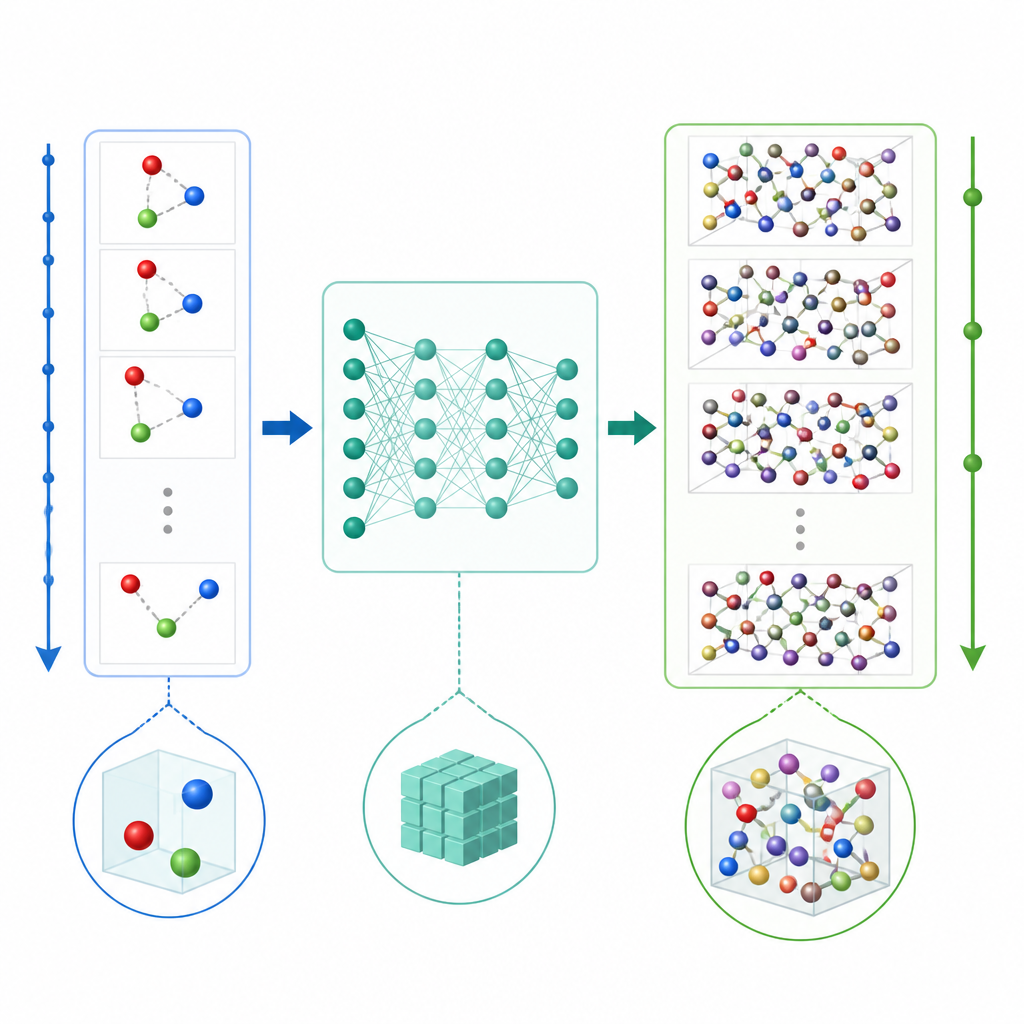
模拟原子的全新捷径
传统的分子动力学通过反复计算原子间的力并以极小的时间步推进位置来更新原子运动,这些时间步往往只有万亿分之一秒的某个分数。即便使用机器学习加速力的计算,微小的时间步依然使得长时程模拟代价高昂。TrajCast 采取不同路径。它不去预测力,而是学会直接从一个完整的系统快照跳到下一个,预测在更大时间间隔后每个原子的位置信息以及运动速度。其内部使用的一类神经网络遵守三维空间的对称性,因此对系统的旋转或平移不会混淆模型。
教 AI 遵守物理规则
要作为物理的可信替代,TrajCast 必须不仅仅画出看起来合理的图像;它还要重现真实原子运动的统计特性。作者用能量和动量守恒的短期准确模拟来训练模型,并将这些守恒定律直接内嵌到网络中,以确保预测的运动在物理上自洽。在长时间的“滚动预测”过程中,TrajCast 重复将自身输出作为下一次输入,同时一个热浴模块温和调整原子速度以控制模拟温度,类似于标准的恒温模拟。该设计使方法能够生成完整轨迹,供科学家提取动力学、结构和能量学性质。
对 TrajCast 的实测检验
团队在三种截然不同的系统上对 TrajCast 进行了基准测试:气相中的单个对乙酰氨基酚(扑热息痛)分子、晶态石英以及室温条件下的液态水。在每种情况下,他们将 AI 生成的轨迹与传统模拟进行比较,侧重于原子如何振动、能量如何分布以及分子如何运动或扩散等度量。对于扑热息痛,TrajCast 使用的时间步比参考模拟大 14 倍,但仍然能很好地重现振动谱、势能分布以及控制分子构象翻转的能垒景观。在石英中,它可以安全地采用比通常大 30 倍的跳跃,同时在纳秒尺度上依然匹配固体的振动特征和能量波动。
从液态水到玻璃态
液态水的挑战更大,因为分子不断形成和断裂氢键。即便如此,在十倍更大的时间步下,TrajCast 仍能反映参考模拟:它捕捉不同频率的振动模态,重现水分子随时间的位移,并给出描述分子相互排列的径向分布函数。作者随后将模型推到远超训练舒适区的情形:使用仅在室温液态水上训练的版本,他们逐步冷却更大的样本以形成超快速淬火得到的玻璃态水——一种具有微妙结构特征的无序固体。值得注意的是,AI 能跟踪扩散如何减慢、结构峰如何变尖以及长程和短程有序性的度量如何演化,这些都与明确包含冷却过程的专门模拟高度一致。
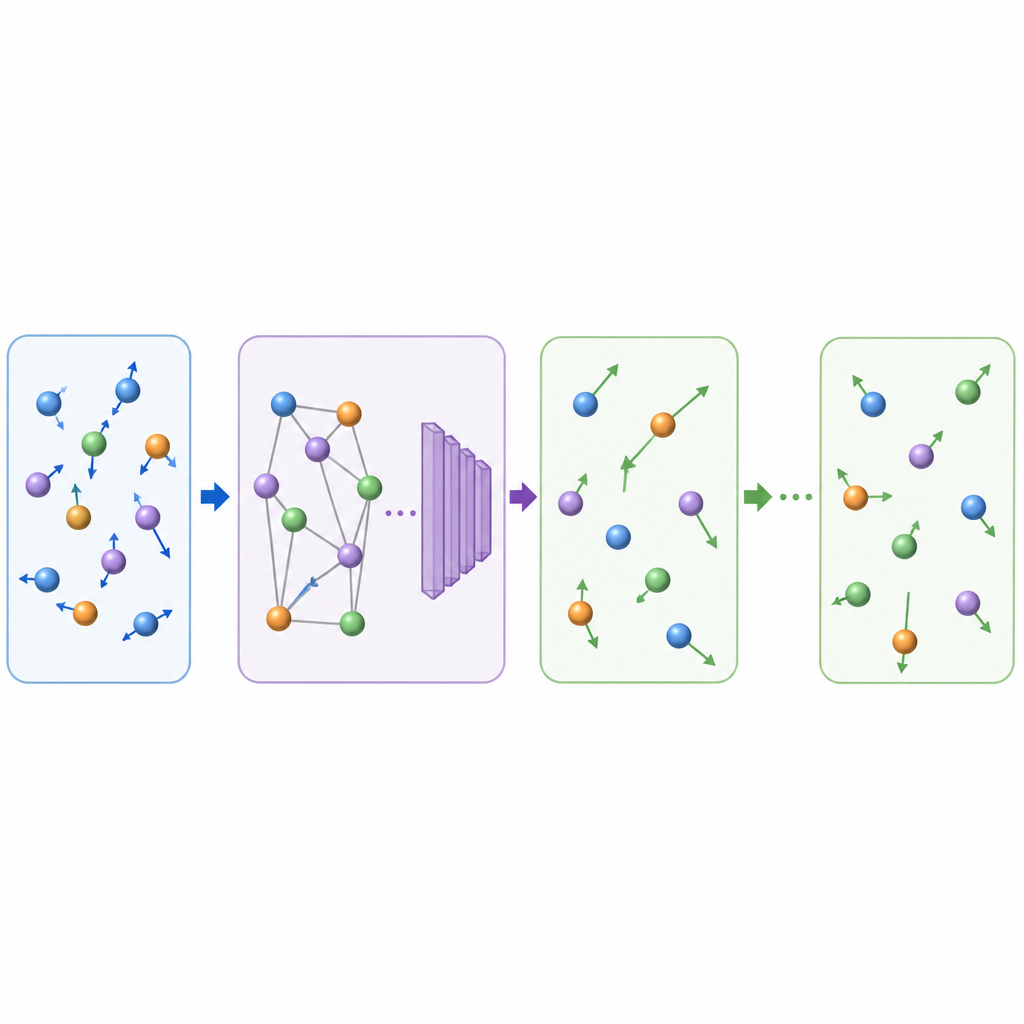
对未来材料发现的意义
通过学习以更大且物理上可信的跳跃推进原子“电影”,TrajCast 能在通常需要模拟更少内容的时间内为含数千个原子的系统生成纳秒级的轨迹数据。该方法保留了足够的细节以恢复关键可观测量,既能处理简单分子也能应对复杂的凝聚相,甚至在无需额外训练的情况下外推到如玻璃形成等新情形。尽管当前版本尚不能预测依赖压强的性质且使用固定的预测步长,但它已展示出一种前景:AI 辅助的模拟有望弥合快速微观运动与塑造现实材料和液体的缓慢过程之间的鸿沟。
引用: Thiemann, F.L., Reschützegger, T., Esposito, M. et al. Force-free molecular dynamics through autoregressive equivariant networks. Nat Mach Intell 8, 764–776 (2026). https://doi.org/10.1038/s42256-026-01227-7
关键词: 分子动力学, 机器学习, 原子尺度模拟, 材料科学, 玻璃态水