Clear Sky Science · it
Dinámica molecolare senza forza tramite reti autoregressive equivarianti
Perché accelerare i "film" atomici è importante
Molte delle questioni più intriganti in chimica, fisica e scienza dei materiali riguardano il modo in cui gli atomi si muovono: come si agita una molecola analgesica, come vibra il quarzo, come l'acqua si congela in strane forme vetrose. Gli esperimenti al computer chiamati dinamica molecolare simulano questi film atomici, ma sono così costosi in termini computazionali che gli scienziati vedono spesso solo una frazione minima dell'azione. Questo articolo presenta TrajCast, un nuovo quadro di intelligenza artificiale che impara ad avanzare velocemente questi film atomici senza sacrificare la fisica essenziale.
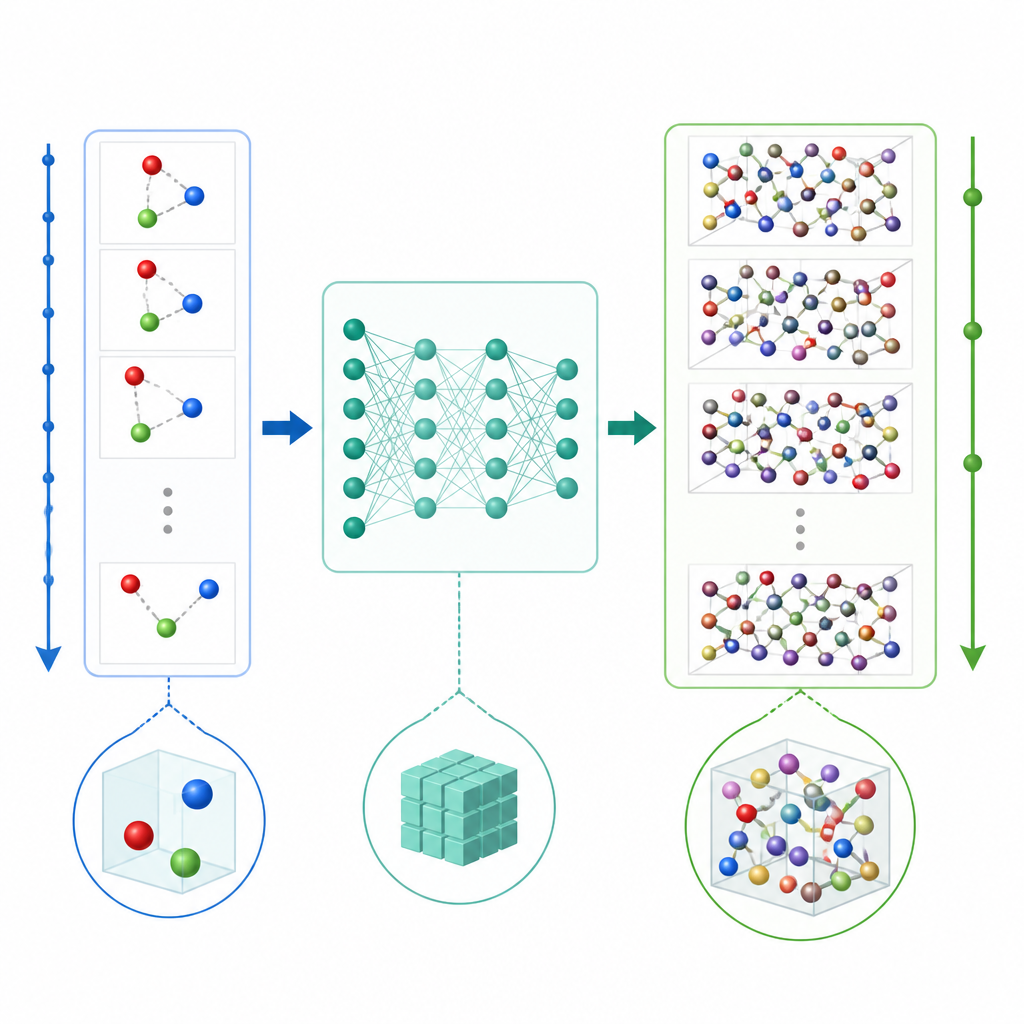
Una nuova scorciatoia per simulare gli atomi
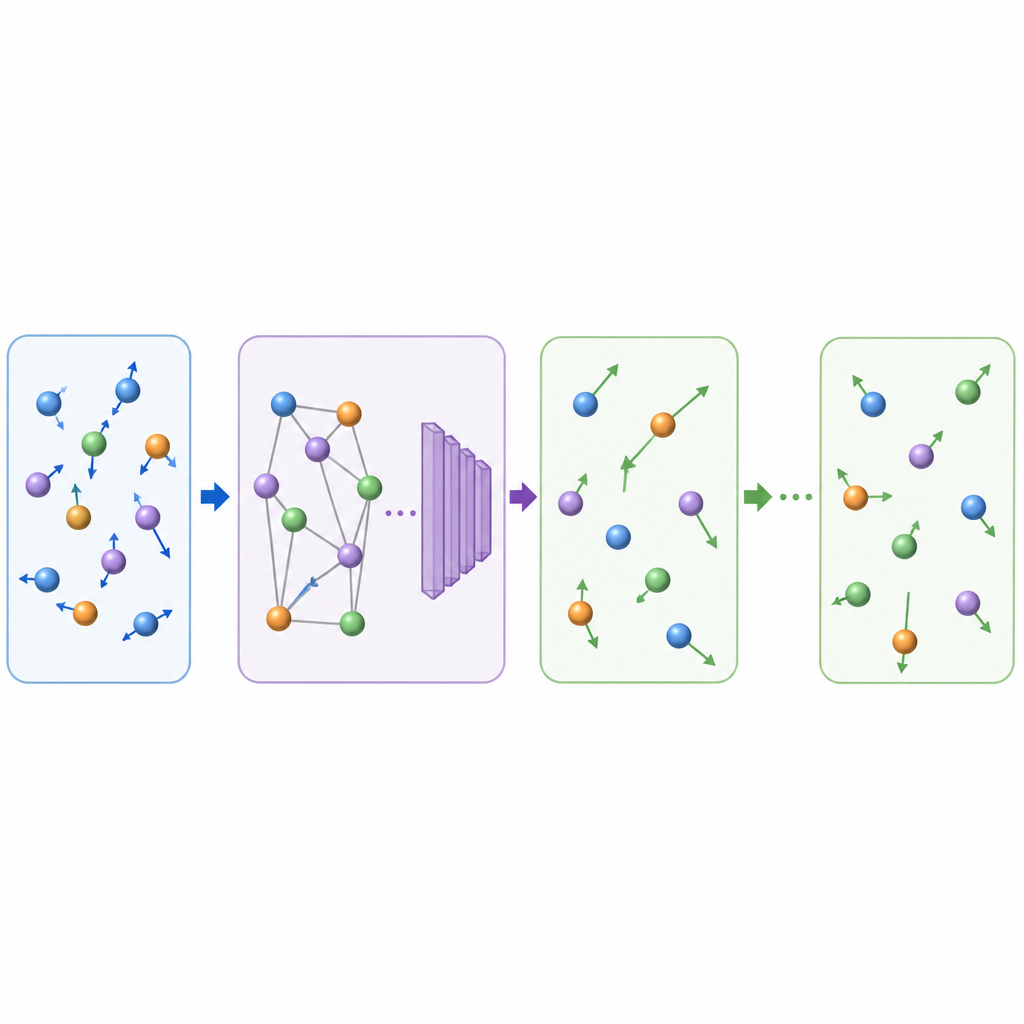
La dinamica molecolare tradizionale aggiorna il moto atomico calcolando ripetutamente le forze tra atomi e poi spostando le posizioni in piccoli intervalli di tempo, spesso una frazione di un trilionesimo di secondo. Anche quando l'apprendimento automatico velocizza i calcoli delle forze, il piccolo passo temporale rende comunque costosi i film lunghi. TrajCast prende una via diversa. Invece di prevedere le forze, impara a saltare direttamente da un'istantanea completa del sistema alla successiva, prevedendo sia dove si troverà ciascun atomo sia quanto velocemente si muoverà dopo un intervallo di tempo molto più ampio. Sotto il cofano, TrajCast usa una classe di reti neurali che rispettano le simmetrie dello spazio tridimensionale, così ruotare o traslare il sistema non confonde il modello.
Insegnare all'IA a rispettare le regole fisiche
Per agire come un sostituto affidabile della fisica, TrajCast deve fare più che disegnare immagini plausibili; deve riprodurre le statistiche del moto atomico reale. Gli autori addestrano il modello su simulazioni brevi e accurate che conservano energia e quantità di moto, e incorporano direttamente queste leggi di conservazione nella rete in modo che i moti predetti rimangano coerenti dal punto di vista fisico. Durante i lunghi "roll‑out", TrajCast prende ripetutamente il proprio output come input successivo, mentre un modulo termostato aggiusta delicatamente le velocità atomiche per mantenere la temperatura simulata sotto controllo, proprio come nelle simulazioni standard a temperatura costante. Questo progetto permette al metodo di generare traiettorie complete da cui gli scienziati possono estrarre proprietà dinamiche, strutturali ed energetiche.
Mettere TrajCast alla prova
Il team valuta TrajCast su tre sistemi molto diversi: una singola molecola di paracetamolo in fase gassosa, quarzo cristallino e acqua liquida a condizioni ambientali. Per ciascun caso, confrontano le traiettorie generate dall'IA con le simulazioni convenzionali, concentrandosi su misure come come gli atomi vibrano, come l'energia è distribuita e come le molecole si muovono o diffondono. Per il paracetamolo, TrajCast usa passi temporali quattordici volte più grandi rispetto alla simulazione di riferimento, eppure riproduce da vicino lo spettro dei modi vibratori, la distribuzione delle energie potenziali e il paesaggio delle barriere energetiche che governano come la molecola si capovolge tra forme diverse. Nel quarzo, può permettersi salti trenta volte più grandi del solito, mantenendo comunque le firme vibratorie e le fluttuazioni energetiche del solido su scale di nanosecondi.
Dall'acqua liquida agli stati vetrosi
L'acqua liquida rappresenta una sfida più dura perché le molecole formano e rompono continuamente legami a idrogeno. Anche qui, con un passo temporale dieci volte maggiore, TrajCast rispecchia le simulazioni di riferimento: cattura i modi vibratori attraverso le frequenze, riproduce quanto lontano viaggiano le molecole d'acqua nel tempo e fornisce funzioni di distribuzione radiale che descrivono come le molecole sono disposte l'una attorno all'altra. Gli autori spingono poi il modello ben oltre la sua zona di comfort di addestramento. Usando una versione addestrata solo sull'acqua liquida a temperatura ambiente, raffreddano gradualmente un campione più grande per formare acqua vetrosa iperraffreddata, un solido disordinato con sottili firme strutturali. Sorprendentemente, l'IA segue come la diffusione rallenti, come i picchi strutturali si affilano e come evolvono le misure di ordine a lungo e a breve raggio, in stretto accordo con simulazioni dedicate che includono esplicitamente il raffreddamento.
Cosa significa per la scoperta di materiali futura
Imparando ad avanzare i film atomici con salti più grandi e fisicamente fedeli, TrajCast può generare nanosecondi di dati di traiettoria per sistemi contenenti migliaia di atomi nel tempo che normalmente servirebbe per simulare molto meno. Il metodo conserva abbastanza dettaglio per recuperare osservabili chiave, gestisce sia molecole semplici sia fasi condensate complesse, e può persino estrapolare a nuovi regimi come la formazione del vetro senza ulteriore addestramento. Sebbene la versione attuale non possa ancora prevedere proprietà dipendenti dalla pressione e usi un passo di previsione fisso, indica già un futuro in cui le simulazioni assistite da IA colmano il divario tra i rapidi moti microscopici e i processi lenti che plasmano materiali e liquidi nel mondo reale.
Citazione: Thiemann, F.L., Reschützegger, T., Esposito, M. et al. Force-free molecular dynamics through autoregressive equivariant networks. Nat Mach Intell 8, 764–776 (2026). https://doi.org/10.1038/s42256-026-01227-7
Parole chiave: dynamica molecolare, apprendimento automatico, simulazione atomistica, scienza dei materiali, acqua vetrosa