Clear Sky Science · en
Force-free molecular dynamics through autoregressive equivariant networks
Why speeding up atomic movies matters
Many of the most intriguing questions in chemistry, physics and materials science come down to how atoms move: how a painkiller molecule wiggles, how quartz vibrates, how water freezes into strange glassy forms. Computer experiments called molecular dynamics simulate these atomic movies, but they are so computationally demanding that scientists often see only a tiny fraction of the action. This article presents TrajCast, a new artificial intelligence framework that learns to fast‑forward these atomic films without sacrificing the essential physics.
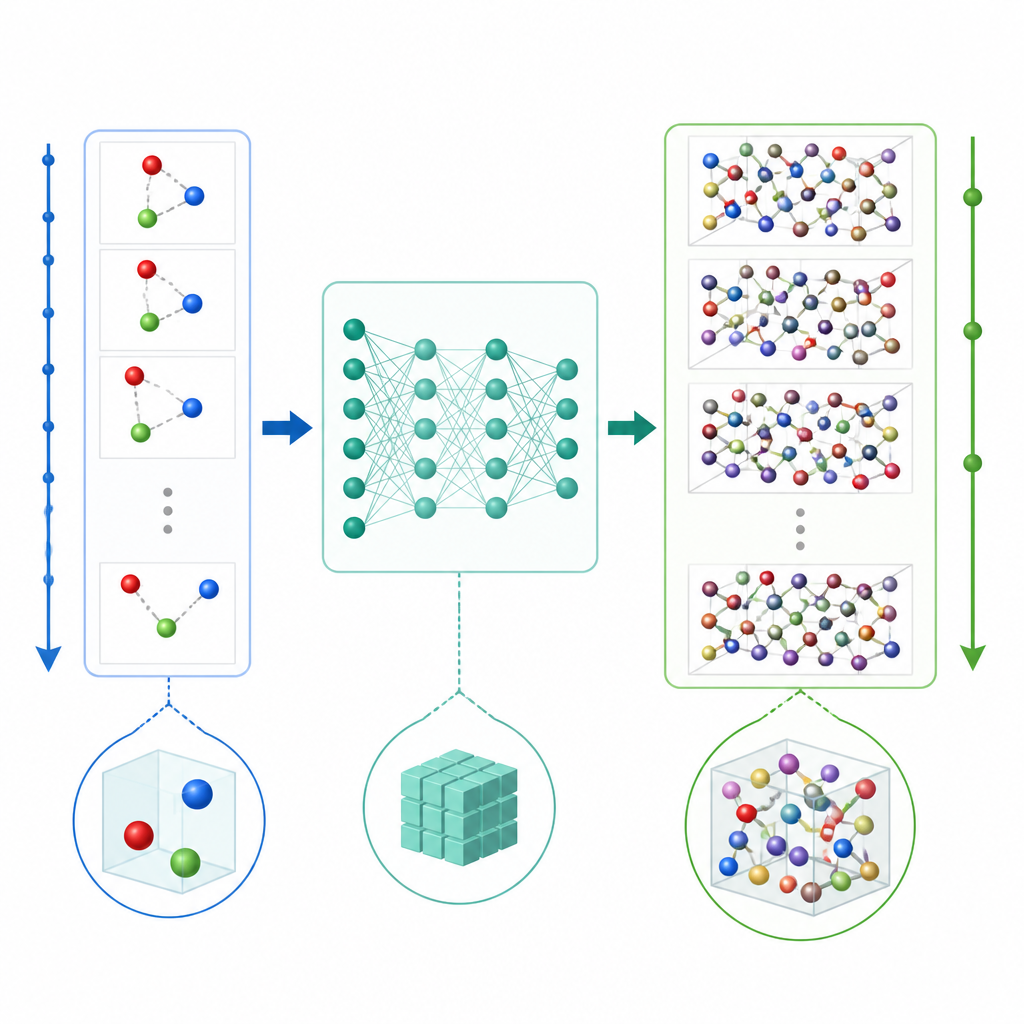
A new shortcut for simulating atoms
Traditional molecular dynamics updates atomic motion by repeatedly computing the forces between atoms and then nudging positions forward in very small time steps, often a fraction of a trillionth of a second. Even when machine learning is used to speed up the force calculations, the tiny time step still makes long movies expensive. TrajCast takes a different route. Instead of predicting forces, it learns to jump directly from one full snapshot of the system to the next, forecasting both where each atom will be and how fast it will be moving after a much larger time interval. Under the hood, TrajCast uses a class of neural networks that respects the symmetries of three‑dimensional space so that rotating or translating the system does not confuse the model.
Teaching AI to respect physical rules
To act as a trustworthy stand‑in for physics, TrajCast must do more than draw plausible pictures; it must reproduce the statistics of real atomic motion. The authors train the model on short, accurate simulations that conserve energy and momentum, and they build these conservation laws directly into the network so that predicted motions remain physically consistent. During long “roll‑outs,” TrajCast repeatedly takes its own output as the next input, while a thermostat module gently adjusts atomic speeds to keep the simulated temperature under control, just as in standard constant‑temperature simulations. This design allows the method to generate full trajectories from which scientists can extract dynamical, structural and energetic properties.
Putting TrajCast to the test
The team benchmarks TrajCast on three very different systems: a single paracetamol molecule in the gas phase, crystalline quartz and liquid water at room conditions. For each case, they compare the AI‑generated trajectories with conventional simulations, focusing on measures such as how atoms vibrate, how energy is distributed and how molecules move or diffuse. For paracetamol, TrajCast uses time steps fourteen times larger than the reference simulation, yet it closely reproduces the spectrum of vibrational motions, the spread of potential energies and the landscape of energy barriers that govern how the molecule flips between shapes. In quartz, it can safely take jumps thirty times larger than usual, while still matching the vibrational signatures and energy fluctuations of the solid over nanoseconds.
From liquid water to glassy states
Liquid water offers a tougher challenge because molecules constantly form and break hydrogen bonds. Even here, with a tenfold larger time step, TrajCast mirrors the reference simulations: it captures vibrational modes across frequencies, reproduces how far water molecules travel over time and yields radial distribution functions that describe how molecules are arranged around one another. The authors then push the model far beyond its training comfort zone. Using a version trained only on room‑temperature liquid water, they gradually cool a larger sample to form hyperquenched glassy water, a disordered solid with subtle structural signatures. Remarkably, the AI tracks how diffusion slows, how structural peaks sharpen and how measures of long‑ and short‑range order evolve, in close agreement with dedicated simulations that explicitly include cooling.
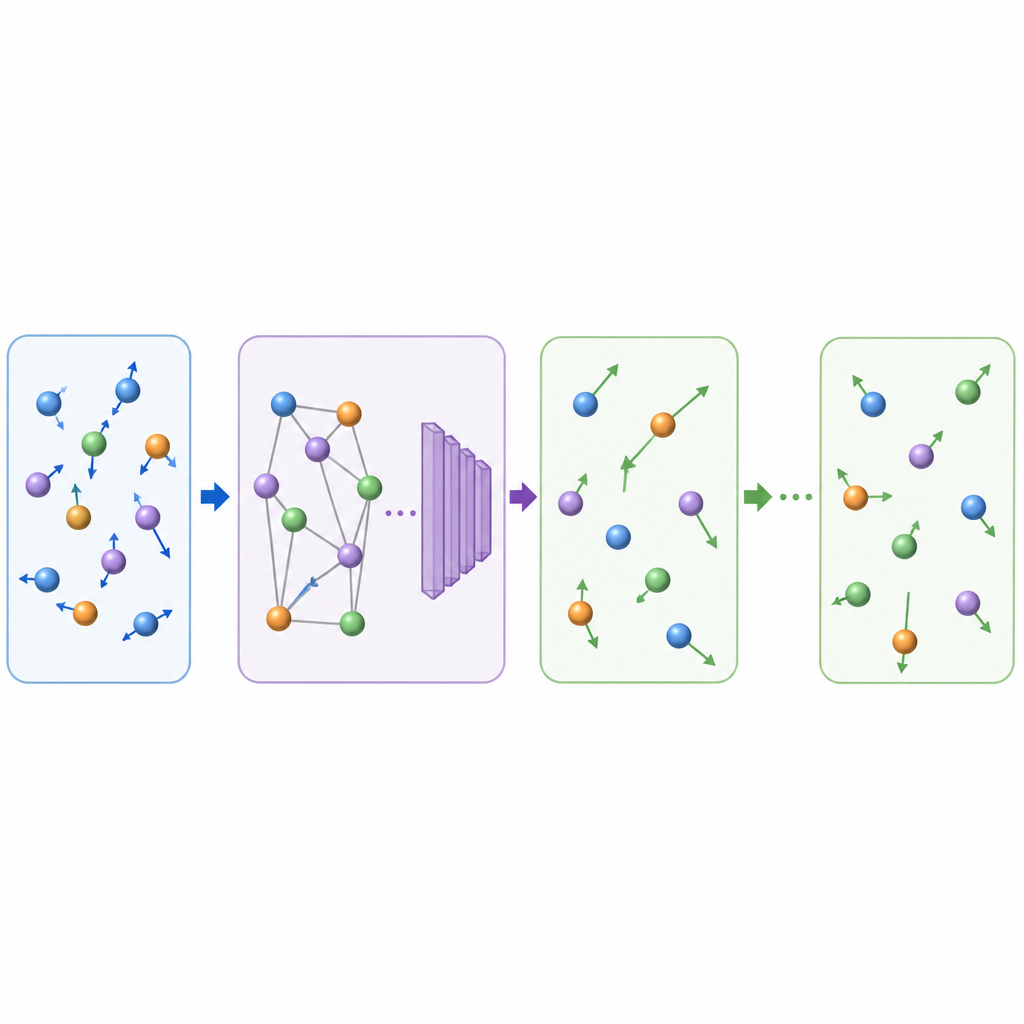
What this means for future materials discovery
By learning to advance atomic movies in larger, physically faithful jumps, TrajCast can generate nanoseconds of trajectory data for systems containing thousands of atoms in the time it would normally take to simulate far less. The method retains enough detail to recover key observables, handles both simple molecules and complex condensed phases, and can even extrapolate to new regimes such as glass formation without additional training. Although the current version cannot yet predict pressure‑dependent properties and uses a fixed forecast step, it already points to a future in which AI‑assisted simulations bridge the gap between fast microscopic motions and the slow processes that shape materials and liquids in the real world.
Citation: Thiemann, F.L., Reschützegger, T., Esposito, M. et al. Force-free molecular dynamics through autoregressive equivariant networks. Nat Mach Intell 8, 764–776 (2026). https://doi.org/10.1038/s42256-026-01227-7
Keywords: molecular dynamics, machine learning, atomistic simulation, materials science, glassy water